
Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification

Abstract
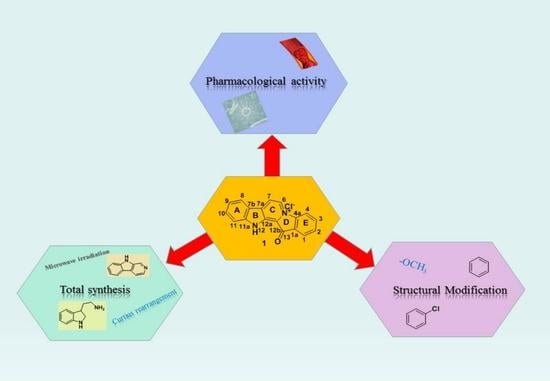
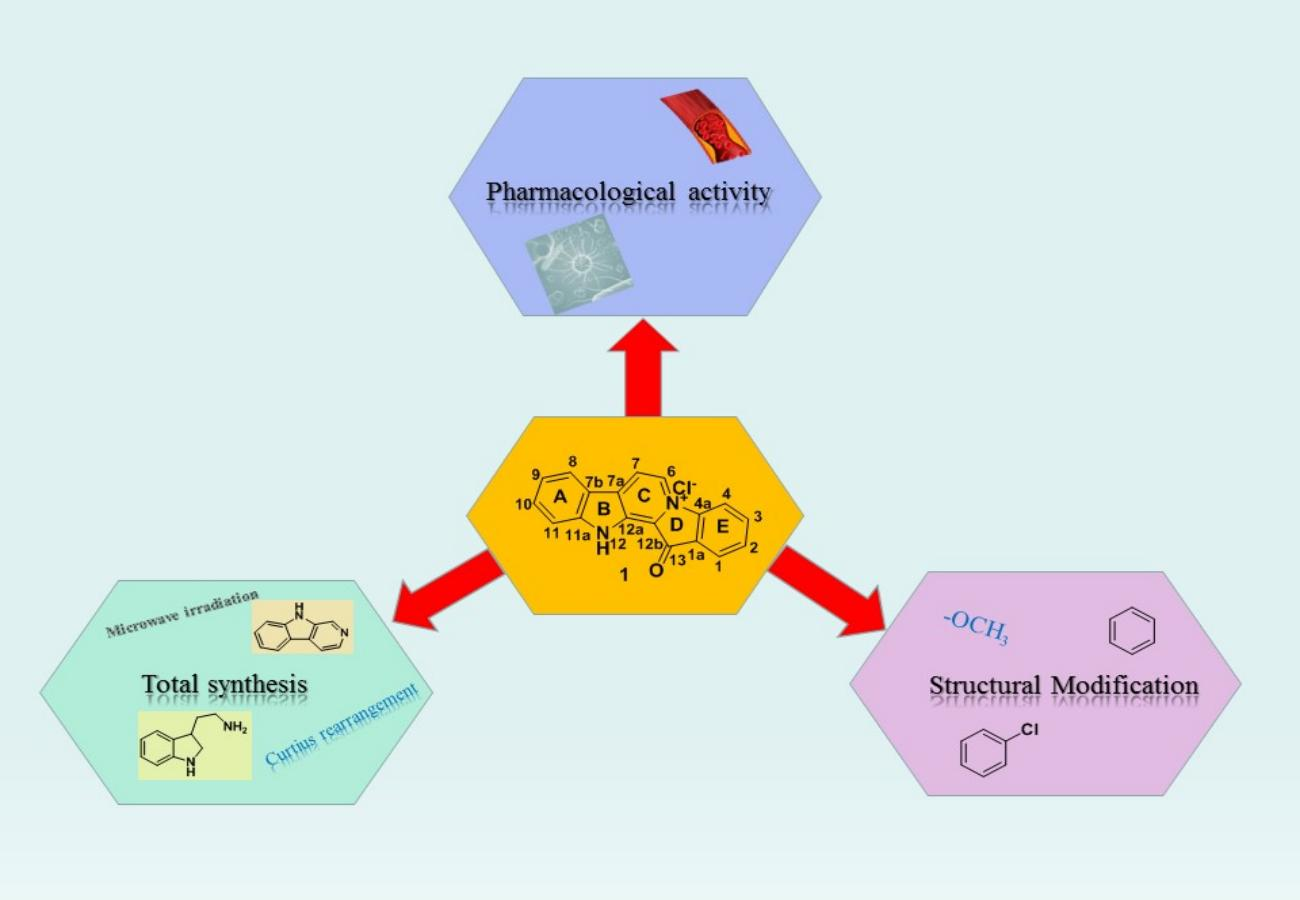
:
1. Introduction
2. Pharmacological Activity
2.1. Anti-Tumor Effects
2.1.1. Anti-Tumor Mechanism
2.1.2. Inhibition of Human Alveolar Rhabdomyosarcoma Cells
2.1.3. Inhibition of Leukemia Cells
2.1.4. Inhibition of Liver Cancer Cells and Human Venous Endothelial Cells
2.1.5. Inhibition of Melanoma
2.1.6. Inhibition of Small Cell Lung Cancer Cells
2.1.7. Inhibition of Ovarian Cancer Cells
2.1.8. Inhibition of Other Tumor Cells
2.1.9. Increasing the Anticancer Effect of Other Drugs
2.2. Analgesic Activity
2.3. Anti-Thrombotic Activity
2.4. Anti-Alzheimer Activity
2.5. Anti-Plasmodial Activity
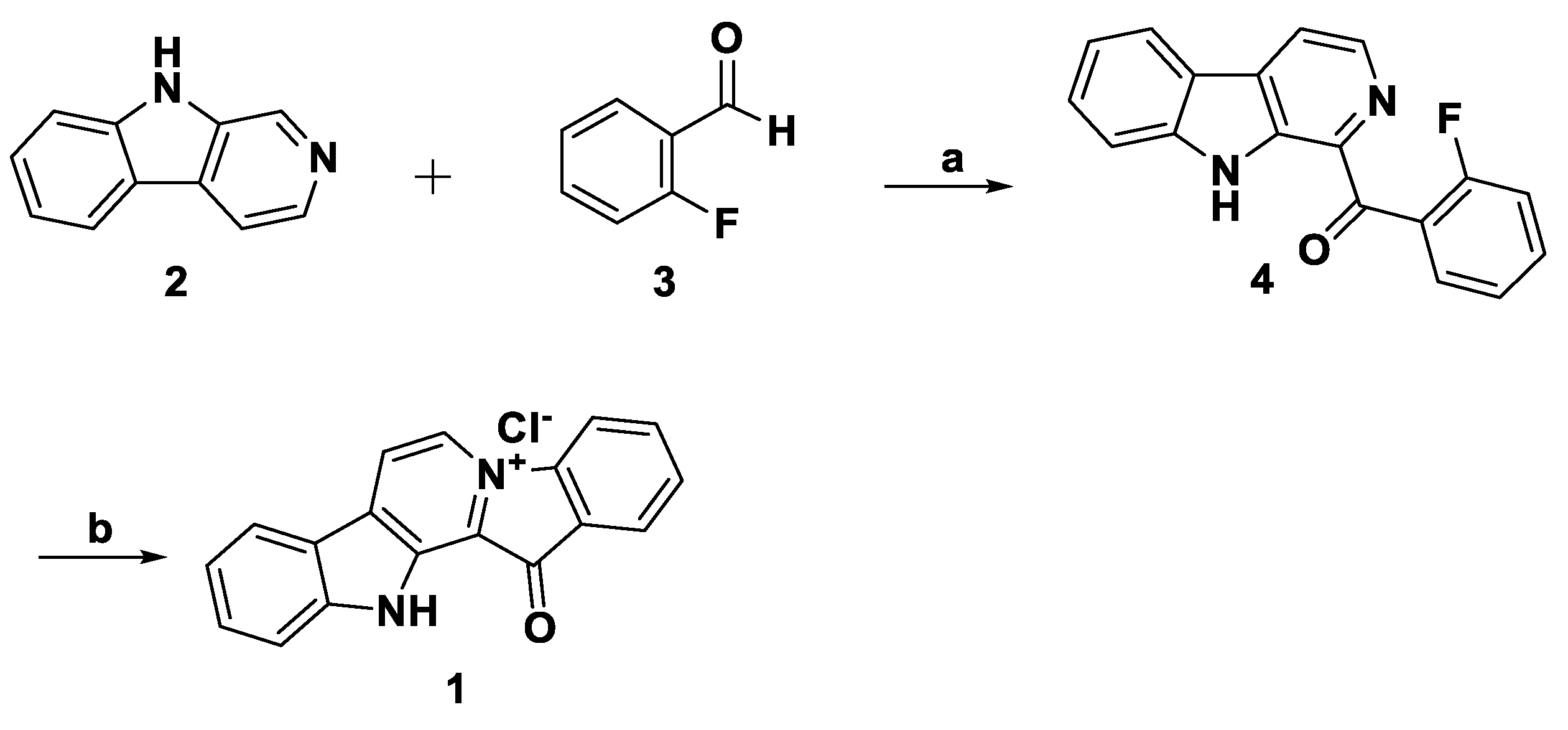
3. Total Synthesis
4. Structural Modification
4.1. Anti-Tumor Fascaplysin Derivatives
4.1.1. One-Ring-Modified Fascaplysin Derivatives
4.1.2. Two-Rings-Modified Fascaplysin Derivatives
4.1.3. Three-Rings-Modified Fascaplysin Derivatives
4.1.4. Four-Rings-Modified Fascaplysin Derivatives
4.2. Anti-Alzheimer Fascaplysin Derivatives
4.3. Antiplasmodial Fascaplysin Derivatives
4.4. Antibacterial Fascaplysin Derivatives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, A.; Naughton, L.M.; Montanchez, I.; Dobson, A.D.W.; Rai, D.K. Current Status and Future Prospects of Marine Natural Products (MNPs) as Antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Hamann, M.T.J.T.L.i.d. Marine natural products and their potential applications as anti-infective agents. Lancet Infect. Dis. 2003, 3, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Novanna, M.; Ethiraj, K.R.; Kannadasan, S. An Overview of Synthesis of Indole Alkaloids and Biological Activities of Secondary Metabolites Isolated from Hyrtios Species. Mini Rev. Med. Chem. 2019, 19, 194–205. [Google Scholar] [CrossRef]
- Dyshlovoy, S.; Honecker, F.J.M.d. Marine Compounds and Cancer: The First Two Decades of XXI Century. Mar. Drugs 2019, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Paterson, I.; Anderson, E.J.S. Chemistry. The renaissance of natural products as drug candidates. Science 2005, 310, 451–453. [Google Scholar] [CrossRef]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Stonik, V.J.A.N. Marine natural products: A way to new drugs. Acta Nat. 2009, 1, 15–25. [Google Scholar] [CrossRef]
- Zotchev, S.B. Alkaloids from Marine Bacteria. Adv. Bot. Res. 2013, 68, 301–333. [Google Scholar] [CrossRef]
- de Oliveira, J.H.; Nascimento, A.M.; Kossuga, M.H.; Cavalcanti, B.C.; Pessoa, C.O.; Moraes, M.O.; Macedo, M.L.; Ferreira, A.G.; Hajdu, E.; Pinheiro, U.S.; et al. Cytotoxic alkylpiperidine alkaloids from the Brazilian marine sponge Pachychalina alcaloidifera. J. Nat. Prod. 2007, 70, 538–543. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Bromo- and iodo-containing alkaloids from marine microorganisms and sponges. Russ. J. Bioorg. Chem. 2002, 28, 196–208. [Google Scholar] [CrossRef]
- Endo, T.; Tsuda, M.; Fromont, J.; Kobayashi, J. Hyrtinadine A, a bis-indole alkaloid from a marine sponge. J. Nat. Prod. 2007, 70, 423–424. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kurimoto, S.; Kobayashi, J. The manzamine alkaloids. Alkaloids Chem. Biol. 2020, 84, 1–124. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Tanaka, N.; Kobayashi, J.; Kashiwada, Y. Agelamasines A and B, diterpene alkaloids from an Okinawan marine sponge Agelas sp. Mar. Drugs 2018, 72, 364–368. [Google Scholar] [CrossRef]
- Lee, S.; Tanaka, N.; Takahashi, S.; Tsuji, D.; Kim, S.; Kojoma, M.; Itoh, K.; Kobayashi, J.; Kashiwada, Y. AgelasAgesasines A and B, Bromopyrrole Alkaloids from Marine Sponges spp. Mar. Drugs 2020, 18, 455. [Google Scholar] [CrossRef] [PubMed]
- Netz, N.; Opatz, T. Marine indole alkaloids. Mar. Drugs 2015, 13, 4814–4914. [Google Scholar] [CrossRef] [Green Version]
- Plodek, A.; Bracher, F. New Perspectives in the Chemistry of Marine Pyridoacridine Alkaloids. Mar. Drugs 2016, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Rane, R.; Sahu, N.; Shah, C.; Karpoormath, R. Marine bromopyrrole alkaloids: Synthesis and diverse medicinal applications. Curr. Top. Med. Chem. 2014, 14, 253–273. [Google Scholar] [CrossRef]
- Bian, C.; Wang, J.; Zhou, X.; Wu, W.; Guo, R.J.C. Biodiversity Recent Advances on Marine Alkaloids from Sponges. Chem. Biodivers. 2020, 17, e2000186. [Google Scholar] [CrossRef]
- De Souza, É.T.; De Lira, D.P.; De Queiroz, A.C.; Silva, D.J.C.d.; De Aquino, A.B.; Campessato Mella, E.A.; Lorenzo, V.P.; De Miranda, G.E.C.; de Araujo-Junior, J.X.; de Oliveira Chaves, M.C. The antinociceptive and anti-inflammatory activities of caulerpin, a bisindole alkaloid isolated from seaweeds of the genus Caulerpa. Mar. Drugs 2009, 7, 689–704. [Google Scholar] [CrossRef] [Green Version]
- Melander, R.; Liu, H.; Stephens, M.; Bewley, C.; Melander, C. Marine sponge alkaloids as a source of anti-bacterial adjuvants. Bioorg. Med. Chem. Lett. 2016, 26, 5863–5866. [Google Scholar] [CrossRef] [Green Version]
- Shubina, L.; Makarieva, T.; von Amsberg, G.; Denisenko, V.; Popov, R.; Dyshlovoy, S. Monanchora pulchraMonanchoxymycalin C with anticancer properties, new analogue of crambescidin 800 from the marine sponge. Nat. Prod. Res. 2019, 33, 1415–1422. [Google Scholar] [CrossRef]
- Souza, C.; Bezerra, W.; Souto, J. Marine Alkaloids with Anti-Inflammatory Activity: Current Knowledge and Future Perspectives. Mar. Drugs 2020, 18, 147. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Xu, Y.; Li, X.; Yao, H.; Lin, K. Development and strategies of CDK4/6 inhibitors. Future Med. Chem. 2020, 12, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, M.; Steinbrecher, T.; Schmid, R.J.P.o. Fascaplysin as a specific inhibitor for CDK4: Insights from molecular modelling. PLoS ONE 2012, 7, e42612. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Guru, S.; Pathania, A.; Manda, S.; Kumar, A.; Bharate, S.; Vishwakarma, R.; Malik, F.; Bhushan, S. Fascaplysin induces caspase mediated crosstalk between apoptosis and autophagy through the inhibition of PI3K/AKT/mTOR signaling cascade in human leukemia HL-60 cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef]
- Liu, L.; Wu, J.; Ong, S.; Chen, T. Cyclin-dependent kinase 4 phosphorylates and positively regulates PAX3-FOXO1 in human alveolar rhabdomyosarcoma cells. PLoS ONE 2013, 8, e58193. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Chen, Y.; Tan, S.; Wu, S.; Huang, Y.; Fu, S.; Luo, F.; He, J. The Research Progress of Antiangiogenic Therapy, Immune Therapy and Tumor Microenvironment. Front. Immunol. 2022, 13, 802846. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Chen, H.; Lu, X.; Wang, F.; Xu, W.; Jin, H.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar] [CrossRef]
- Zhidkov, M.; Kaune, M.; Kantemirov, A.; Smirnova, P.; Spirin, P.; Sidorova, M.; Stadnik, S.; Shyrokova, E.; Kaluzhny, D.; Tryapkin, O.; et al. Study of Structure-Activity Relationships of the Marine Alkaloid Fascaplysin and Its Derivatives as Potent Anticancer Agents. Mar. Drugs 2022, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Huang, W.; Zhu, H.; Peng, C.; Zhao, Q.; Han, B. Advances in indole-containing alkaloids as potential anticancer agents by regulating autophagy. Biomed. Pharmacother. 2022, 149, 112827. [Google Scholar] [CrossRef]
- Wang, F.; Chen, H.; Yan, X.; Zheng, Y. Fascaplysin sensitizes cells to TRAIL-induced apoptosis through upregulating DR5 expression. Chin. J. Oceanol. Limnol. 2013, 31, 560–569. [Google Scholar] [CrossRef]
- Meng, N.; Mu, X.; Lv, X.; Wang, L.; Li, N.; Gong, Y. Autophagy represses fascaplysin-induced apoptosis and angiogenesis inhibition via ROS and p8 in vascular endothelia cells. Biomed. Pharmacother. 2019, 114, 108866. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lu, X.; Lin, J.; Chen, H.; Yan, X.; Wang, F.; Xu, W. Direct effects of fascaplysin on human umbilical vein endothelial cells attributing the anti-angiogenesis activity. Biomed. Pharmacother. 2010, 64, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yan, X.; Chen, H.J.C.c. pharmacology Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol. 2007, 59, 439–445. [Google Scholar] [CrossRef]
- Oh, T.; Lee, Y.; Nam, T.; Ko, Y.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.; Kim, Y.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. [Google Scholar] [CrossRef]
- Mahgoub, T.; Eustace, A.; Collins, D.; Walsh, N.; O’Donovan, N.; Crown, J. Kinase inhibitor screening identifies CDK4 as a potential therapeutic target for melanoma. Int. J. Oncol. 2015, 47, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G. Cytotoxic effects of fascaplysin against small cell lung cancer cell lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef] [Green Version]
- Rath, B.; Hochmair, M.; Plangger, A.; Hamilton, G. Anticancer Activity of Fascaplysin against Lung Cancer Cell and Small Cell Lung Cancer Circulating Tumor Cell Lines. Mar. Drugs 2018, 16, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plangger, A.; Rath, B.; Hochmair, M.; Funovics, M.; Neumayer, C.; Zeillinger, R.; Hamilton, G. Synergistic cytotoxicity of the CDK4 inhibitor Fascaplysin in combination with EGFR inhibitor Afatinib against Non-small Cell Lung Cancer. Investig. New Drugs 2022, 40, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Xu, G. Fascaplysin Induces Apoptosis and Ferroptosis, and Enhances Anti-PD-1 Immunotherapy in Non-Small Cell Lung Cancer (NSCLC) by Promoting PD-L1 Expression. Int. J. Mol. Sci. 2022, 23, 13774. [Google Scholar] [CrossRef]
- Chen, S.; Guan, X.; Wang, L.; Li, B.; Sang, X.; Liu, Y.; Zhao, Y. Fascaplysin inhibit ovarian cancer cell proliferation and metastasis through inhibiting CDK4. Gene 2017, 635, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lyakhova, I.; Piatkova, M.; Gulaia, V.; Romanishin, A.; Shmelev, M.; Bryukhovetskiy, A.; Sharma, A.; Sharma, H.; Khotimchenko, R.; Bryukhovetskiy, I.J.I.r.o.n. Alkaloids of fascaplysin are promising chemotherapeutic agents for the treatment of glioblastoma: Review. Int. Rev. Neurobiol. 2020, 151, 299–324. [Google Scholar] [CrossRef]
- Bryukhovetskiy, I.; Lyakhova, I.; Mischenko, P.; Milkina, E.; Zaitsev, S.; Khotimchenko, Y.; Bryukhovetskiy, A.; Polevshchikov, A.; Kudryavtsev, I.; Khotimchenko, M. Alkaloids of fascaplysin are effective conventional chemotherapeutic drugs, inhibiting the proliferation of C6 glioma cells and causing their death in vitro. Oncol. Lett. 2017, 13, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Khokhar, S.; Feng, Y.; Campitelli, M.; Ekins, M.; Hooper, J.; Beattie, K.; Sadowski, M.; Nelson, C.; Davis, R. Isolation, structure determination and cytotoxicity studies of tryptophan alkaloids from an Australian marine sponge Hyrtios sp. Bioorg. Med. Chem. Lett. 2014, 24, 3329–3332. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.-I.; Lee, J.H.; Kim, S.; Nam, T.-J.; Kim, Y.-S.; Kim, B.M.; Yim, W.J.; Lim, J.-H. Fascaplysin sensitizes anti-cancer effects of drugs targeting AKT and AMPK. Molecules 2017, 23, 42. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.; Milan-Lobo, L.; Che, T.; Ferwerda, M.; Lambu, E.; McIntosh, N.; Li, F.; He, L.; Lorig-Roach, N.; Crews, P.; et al. dentification of the First Marine-Derived Opioid Receptor “Balanced“ Agonist with a Signaling Profile That Resembles the Endorphins. ACS Chem. Neurosci. 2017, 8, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Ampofo, E.; Später, T.; Müller, I.; Eichler, H.; Menger, M.D.; Laschke, M.W. The marine-derived kinase inhibitor fascaplysin exerts anti-thrombotic activity. Mar. Drugs 2015, 13, 6774–6791. [Google Scholar] [CrossRef] [Green Version]
- Manda, S.; Sharma, S.; Wani, A.; Joshi, P.; Kumar, V.; Guru, S.K.; Bharate, S.S.; Bhushan, S.; Vishwakarma, R.A.; Kumar, A. Discovery of a marine-derived bis-indole alkaloid fascaplysin, as a new class of potent P-glycoprotein inducer and establishment of its structure–activity relationship. Eur. J. Med. Chem. 2016, 107, 1–11. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef]
- Ryan, K.S.; Drennan, C.L. Divergent pathways in the biosynthesis of bisindole natural products. Chem. Biol. 2009, 16, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and biology of fascaplysin, a potent marine-derived CDK-4 inhibitor. Mini Rev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Kaminskii, V.A. A new method for the synthesis of the marine alkaloid fascaplysin based on the microwave-assisted Minisci reaction. Tetrahedron Lett. 2013, 54, 3530–3532. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R.A. Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. MedChemComm 2012, 3, 1098–1103. [Google Scholar] [CrossRef]
- Battini, N.; Padala, A.K.; Mupparapu, N.; Vishwakarma, R.A.; Ahmed, Q.N. Unexplored reactivity of 2-oxoaldehydes towards Pictet–Spengler conditions: Concise approach to β-carboline based marine natural products. RSC Adv. 2014, 4, 26258–26263. [Google Scholar] [CrossRef]
- Dighe, S.U.; Samanta, S.K.; Kolle, S.; Batra, S. Iodine-mediated oxidative Pictet-Spengler reaction using terminal alkyne as the 2-oxoaldehyde surrogate for the synthesis of 1-aroyl-β-carbolines and fused-nitrogen heterocycles. Tetrahedron 2017, 73, 2455–2467. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Palani, V.; Perea, M.A.; Gardner, K.E.; Sarpong, R. A pyrone remodeling strategy to access diverse heterocycles: Application to the synthesis of fascaplysin natural products. Chem. Sci. 2021, 12, 1528–1534. [Google Scholar] [CrossRef]
- Waldmann, H.; Eberhardt, L.; Wittstein, K.; Kumar, K. Silver catalyzed cascade synthesis of alkaloid ring systems: Concise total synthesis of fascaplysin, homofascaplysin C and analogues. Chem. Commun. 2010, 46, 4622–4624. [Google Scholar] [CrossRef]
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A marine sponge alkaloid derivative 4-chloro fascaplysin inhibits tumor growth and VEGF mediated angiogenesis by disrupting PI3K/Akt/mTOR signaling cascade. Chem. Biol. Interact. 2017, 275, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Lyakhova, I.A.; Bryukhovetsky, I.S.; Kudryavtsev, I.V.; Khotimchenko, Y.S.; Zhidkov, M.E.; Kantemirov, A.V. Antitumor Activity of Fascaplysin Derivatives on Glioblastoma Model In Vitro. Bull. Exp. Biol. Med. 2018, 164, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.; Smirnova, P.; Tryapkin, O.; Kantemirov, A.; Khudyakova, Y.; Malyarenko, O.; Ermakova, S.; Grigorchuk, V.; Kaune, M.; Amsberg, G.; et al. Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues. Mar. Drugs 2019, 17, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyakhova, I.; Piatkova, M.; Khotimchenko, Y.; Zhidkov, M.; Kantemirov, A.; Khotimchenko, R.; Bryukhovetskiy, A.; Sharma, A.; Sharma, H.S.; Bryukhovetskiy, I. 3-Bromofascaplysin is a prospective chemical compound for developing new chemotherapy agents in glioblastoma treatment. Int. Rev. Neurobiol. 2020, 151, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Shakoori, A.; Bremner, J.B.; Willis, A.C.; Haritakun, R.; Keller, P.A. Rapid cascade synthesis of poly-heterocyclic architectures from indigo. J. Org. Chem. 2013, 78, 7639–7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhidkov, M.E.; Sidorova, M.A.; Lyakhova, I.A. One-step transformation of the marine alkaloid fascaplysin into homofascaplysins B and B-1. The first syntheses of 3-bromohomofascaplysin B and 3–bromohomofascaplysin B-1. Tetrahedron Lett. 2018, 59, 1417–1420. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Kaune, M.; Hauschild, J.; Kriegs, M.; Hoffer, K.; Busenbender, T.; Smirnova, P.A.; Zhidkov, M.E.; Poverennaya, E.V.; Oh-Hohenhorst, S.J.; et al. Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells. Mar. Drugs 2020, 18, 609. [Google Scholar] [CrossRef] [PubMed]
- Spirin, P.; Shyrokova, E.; Lebedev, T.; Vagapova, E.; Smirnova, P.; Kantemirov, A.; Dyshlovoy, S.A.; Amsberg, G.V.; Zhidkov, M.; Prassolov, V. Cytotoxic Marine Alkaloid 3,10-Dibromofascaplysin Induces Apoptosis and Synergizes with Cytarabine Resulting in Leukemia Cell Death. Mar. Drugs 2021, 19, 489. [Google Scholar] [CrossRef] [PubMed]
- Mahale, S.; Bharate, S.; Manda, S.; Joshi, P.; Bharate, S.; Jenkins, P.; Vishwakarma, R.; Chaudhuri, B. Biphenyl-4-carboxylic acid [2-(1H-indol-3-yl)-ethyl]-methylamide (CA224), a nonplanar analogue of fascaplysin, inhibits Cdk4 and tubulin polymerization: Evaluation of in vitro and in vivo anticancer activity. J. Med. Chem. 2014, 57, 9658–9672. [Google Scholar] [CrossRef] [PubMed]
- Mahale, S.; Aubry, C.; James Wilson, A.; Jenkins, P.; Maréchal, J.; Sutcliffe, M.; Chaudhuri, B. CA224, a non-planar analogue of fascaplysin, inhibits Cdk4 but not Cdk2 and arrests cells at G0/G1 inhibiting pRB phosphorylation. Bioorg. Med. Chem. Lett. 2006, 16, 4272–4278. [Google Scholar] [CrossRef] [PubMed]
- Mahale, S.; Bharate, S.; Manda, S.; Joshi, P.; Jenkins, P.; Vishwakarma, R.; Chaudhuri, B. disease Antitumour potential of BPT: A dual inhibitor of cdk4 and tubulin polymerization. Cell Death Dis. 2015, 6, e1743. [Google Scholar] [CrossRef] [Green Version]
- Mahale, S.; Aubry, C.; Jenkins, P.; Maréchal, J.; Sutcliffe, M.; Chaudhuri, B. Inhibition of cancer cell growth by cyclin dependent kinase 4 inhibitors synthesized based on the structure of fascaplysin. Bioorg. Chem. 2006, 34, 287–297. [Google Scholar] [CrossRef]
- Liang, Y.; Quan, H.; Bu, T.; Li, X.; Liu, X.; Wang, S.; He, D.; Jia, Q.; Zhang, Y. Comparison of the Inhibitory Binding Modes Between the Planar Fascaplysin and Its Nonplanar Tetrahydro-β-carboline Analogs in CDK4. Front. Chem. 2021, 9, 614154. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, X.; Chen, W.; Ma, Q.; Li, M.; Fan, W.; Zhang, J.; Guo, L. Multicomponent synthesis of novel β-carboline-fused imidazolium derivatives via the Mannich reaction: Cytotoxicity, molecular docking, and mechanistic studies as angiogenesis inhibitors. New J. Chem. 2022, 46, 4427–4435. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, F.; Sang, J.; Lin, M.; Ma, J.; Xiao, X.; Yan, S.; Naman, C.B.; Wang, N.; He, S. 9-Methylfascaplysin is a more potent Aβ aggregation inhibitor than the marine-derived alkaloid, fascaplysin, and produces nanomolar neuroprotective effects in SH-SY5Y cells. Mar. Drugs 2019, 17, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Qiu, H.; Zhang, K.; Zhang, P.; Liang, W.; Yang, M.; Mou, C.; Lin, M.; He, M.; Xiao, X. Fascaplysin derivatives are potent multitarget agents against Alzheimer’s disease: In vitro and in vivo evidence. ACS Chem. Neurosci. 2019, 10, 4741–4756. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ding, Y.; Li, X.; Djigbenou, D.; Grimberg, B.; Ferreira, D.; Ireland, C.; Van Wagoner, R. 3-bromohomofascaplysin A, a fascaplysin analogue from a Fijian Didemnum sp. ascidian. Bioorg. Med. Chem. 2011, 19, 6604–6607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Qiu, H.; Yang, N.; Xie, H.; Liang, W.; Lin, J.; Zhu, H.; Zhou, Y.; Wang, N.; Tan, X.; et al. Fascaplysin derivatives binding to DNA via unique cationic five-ring coplanar backbone showed potent antimicrobial/antibiofilm activity against MRSA in vitro and in vivo. Eur. J. Med. Chem. 2022, 230, 114099. [Google Scholar] [CrossRef]
- Liang, H.; Jiang, Y.; Qiu, H.; Liang, W.; Lin, J.; Lin, J.; Liu, W.; Wang, X.; Cui, W.; Chen, X.; et al. Derivatization of Marine-Derived Fascaplysin via Highly Regioselective Suzuki-Miyaura Coupling Contributing to the Enhanced Antibacterial Activity. ChemistrySelect 2022, 7, e202201441. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | IC50 Value |
|---|---|---|
| Fascaplysin | HL-60 | 0.5 μM |
| Sk-Mel-28 | 0.03–0.22 μM | |
| SCLC | 0.89 μM | |
| CTCs | 0.57 μM | |
| NSCLC | 1.15 μM | |
| LNCaP | 0.54 μM | |
| AChE | 1.49 μM | |
| Hela | 0.56 ± 0.05 μM | |
| P. falciparum strain K1 | 50 ng/mL | |
| P. falciparum strain NF54 | 34 ng/mL | |
| Compound 18 | MDA-MB-231 | 0.3 μM |
| Compound 19 | Sk-Mel-28 | 1.1–1.9 μM |
| Compound 23 | antiplasmodial | 1.1 μM |
| Compound 24 | antiplasmodial | 0.85 μM |
| Compound 30 | CDK4-D1 | 5.5 μM |
| Compound 32 | CDK4-D1 | 20 μM |
| CKD2 | >500 μM | |
| Compound 33 | Hela | 1.03 ± 0.19 μM |
| normal cell lines | 99.82 ± 9.97–429.00 ± 38.87 μM | |
| Compound 55 | ring stage parasites | 0.55 ± 0.11 nM |
| all live parasites | 105 ± 38 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Wang, S.; Li, H.; Hou, Y.; Cao, H.; Hua, H.; Li, D. Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification. Mar. Drugs 2023, 21, 226. https://doi.org/10.3390/md21040226
Wang C, Wang S, Li H, Hou Y, Cao H, Hua H, Li D. Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification. Marine Drugs. 2023; 21(4):226. https://doi.org/10.3390/md21040226
Chicago/Turabian StyleWang, Chao, Siyuan Wang, Haonan Li, Yonglian Hou, Hao Cao, Huiming Hua, and Dahong Li. 2023. "Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification" Marine Drugs 21, no. 4: 226. https://doi.org/10.3390/md21040226
APA StyleWang, C., Wang, S., Li, H., Hou, Y., Cao, H., Hua, H., & Li, D. (2023). Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification. Marine Drugs, 21(4), 226. https://doi.org/10.3390/md21040226