
Substitution of D-Arginine at Position 11 of α-RgIA Potently Inhibits α7 Nicotinic Acetylcholine Receptor

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
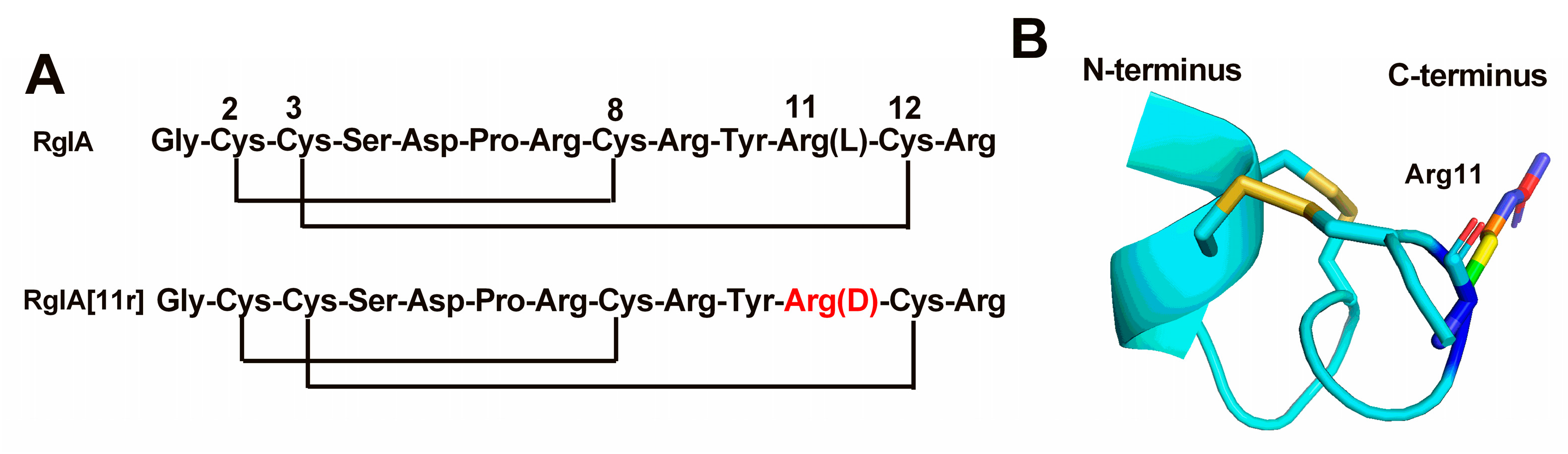
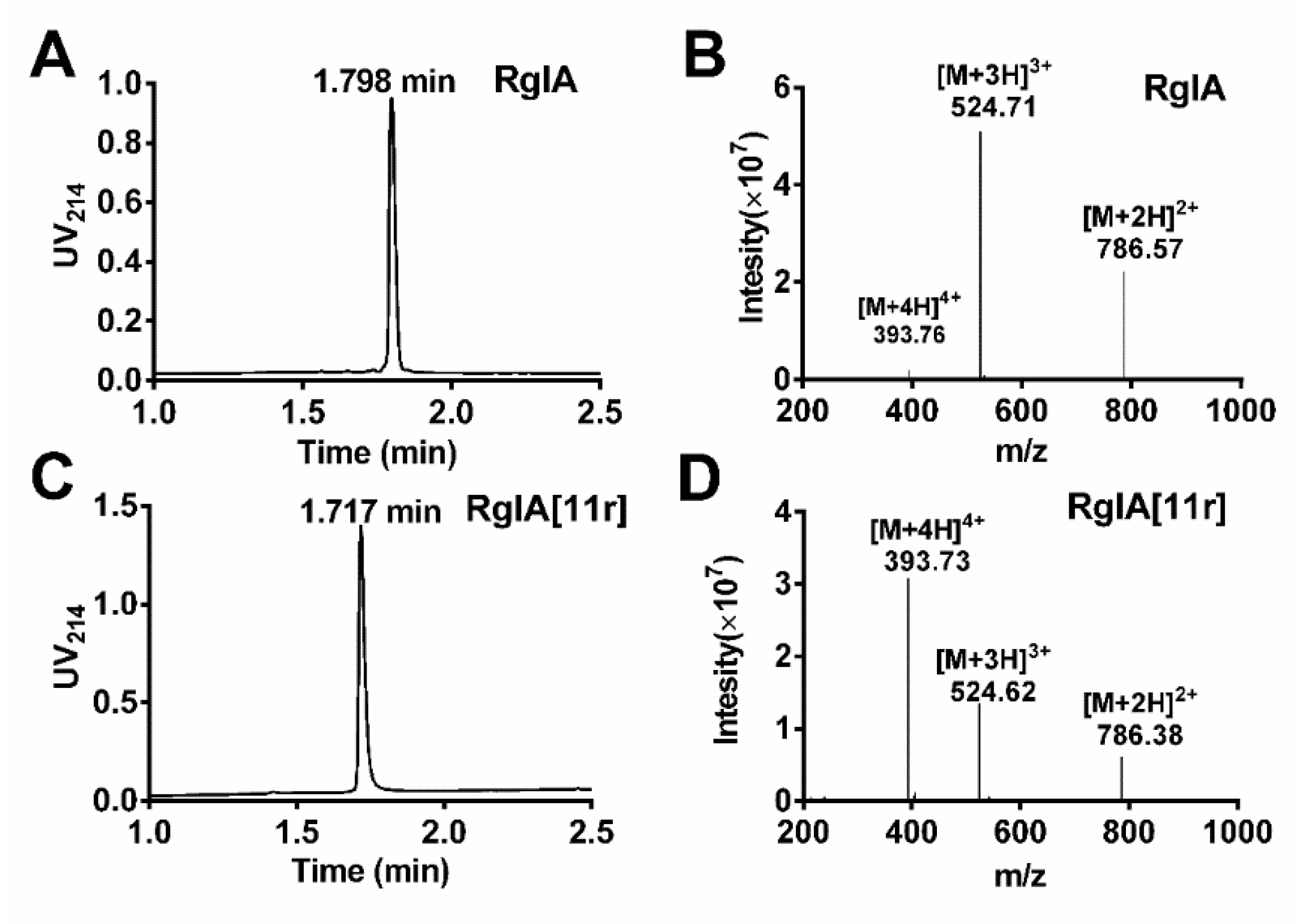
2.1. Synthesis of RgIA and RgIA[11r]
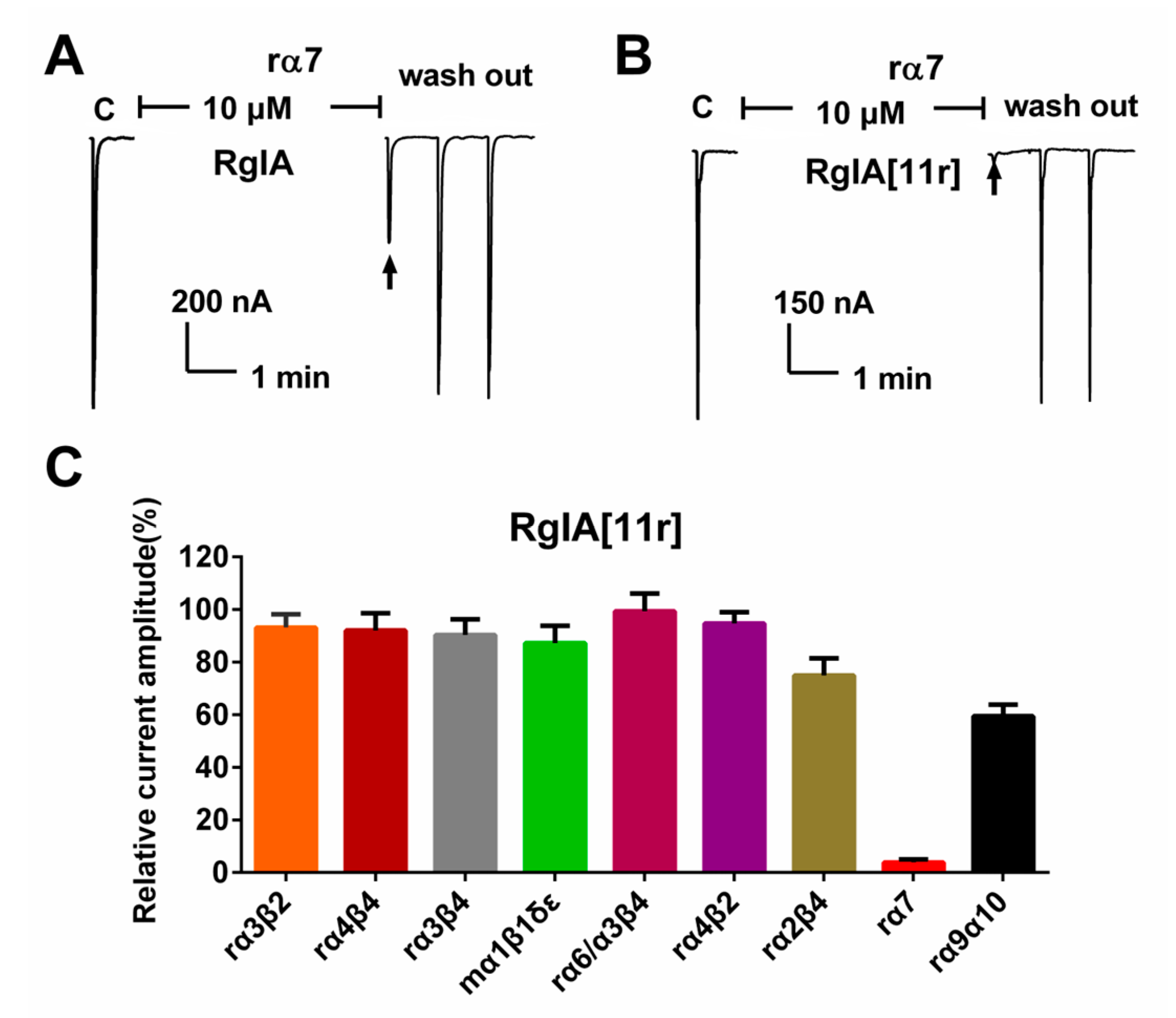
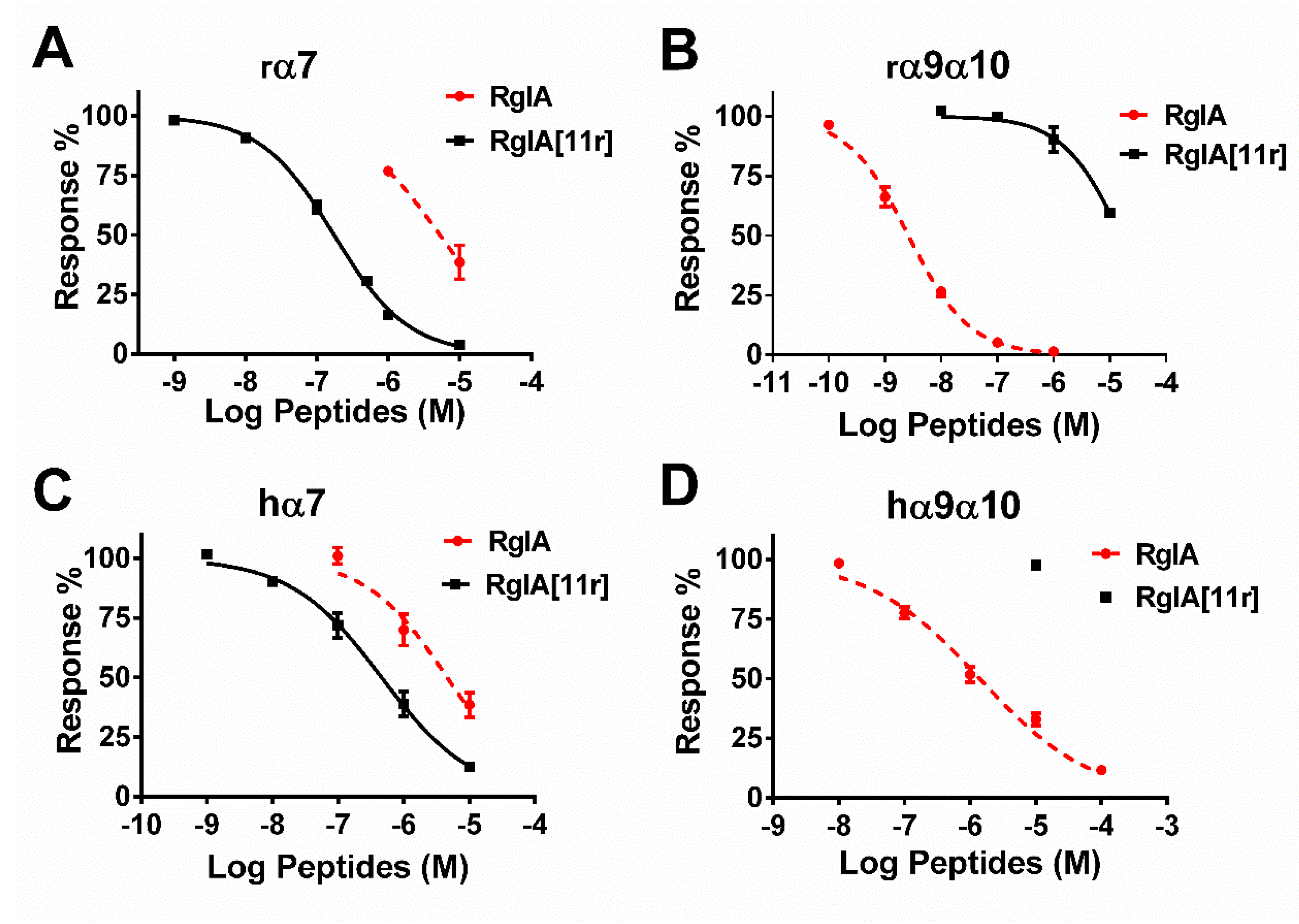
2.2. Potency of RgIA and RgIA[11r] at the Different Types of nAChRs
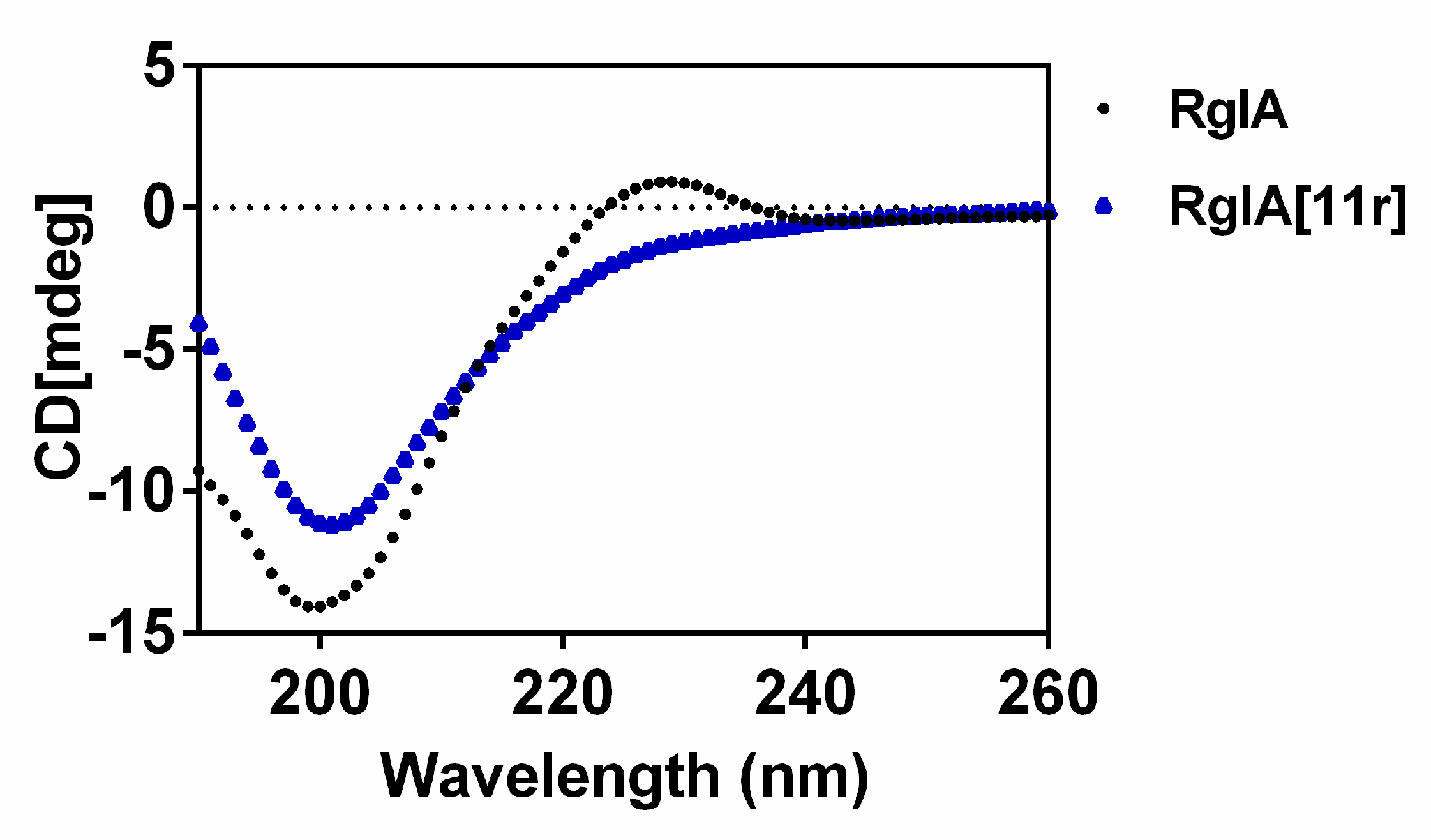
2.3. Circular Dichroism (CD) Spectra of RgIA and RgIA[11r]
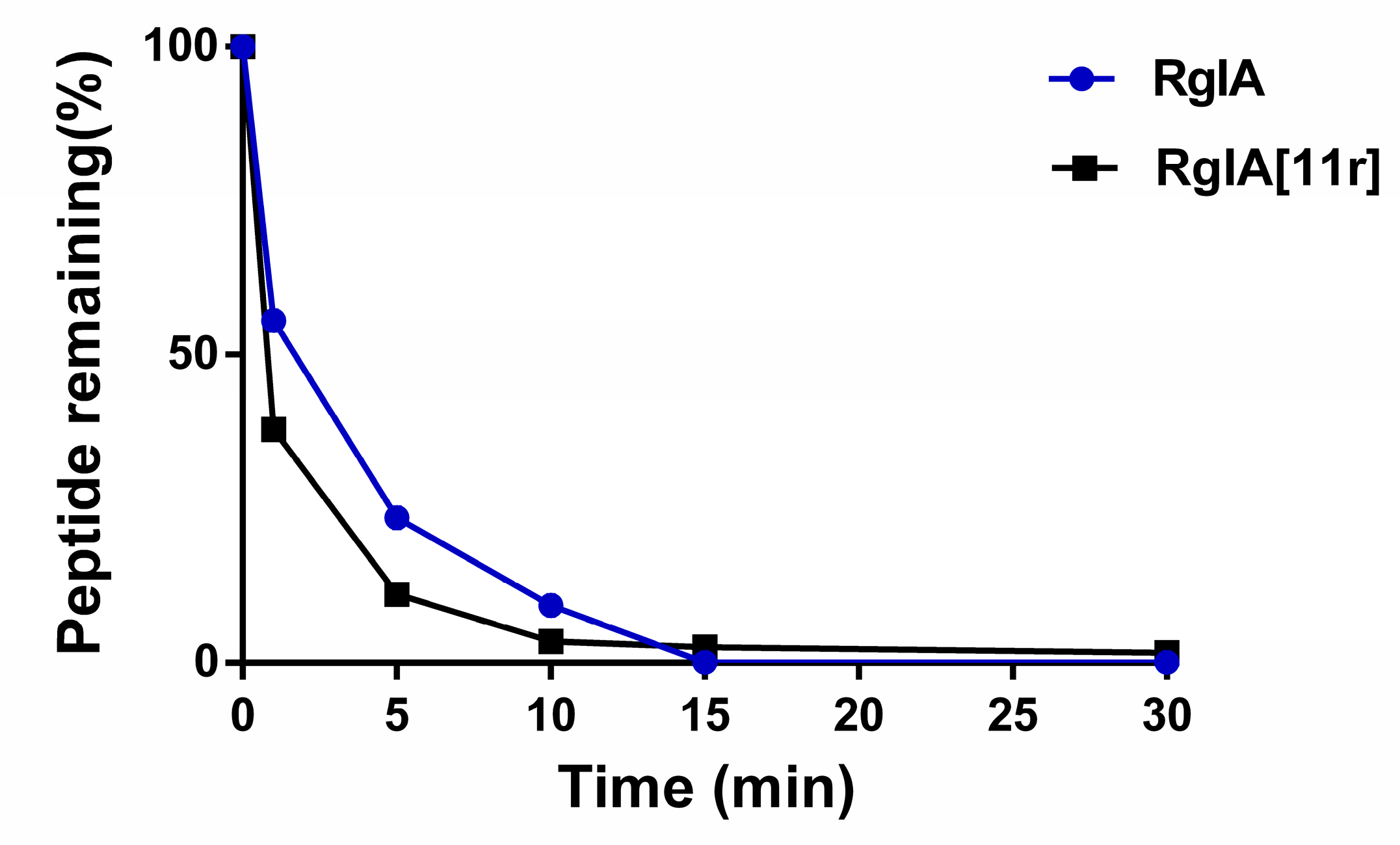
2.4. Serum Stability of RgIA and RgIA[11r]
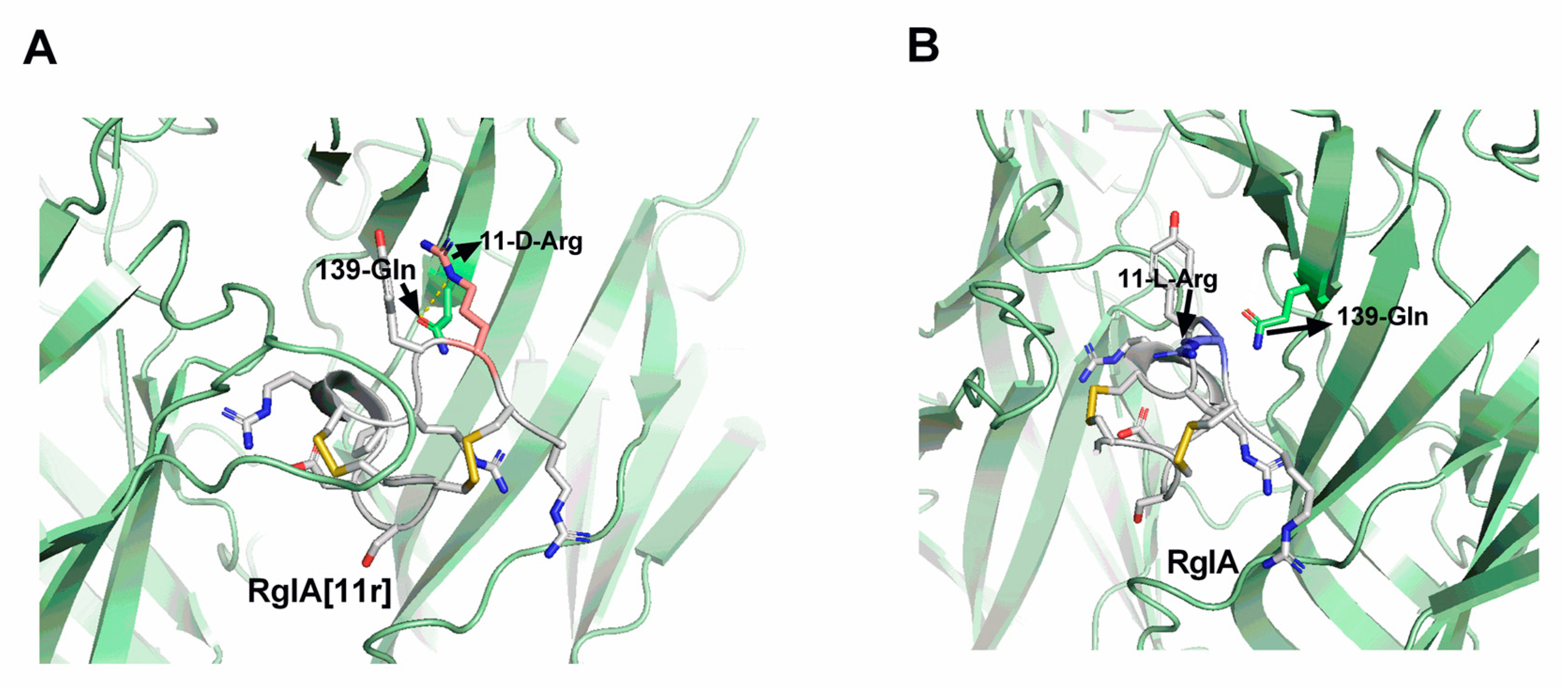
2.5. Molecular Docking (MD) Demonstrates Altered Potency between RgIA[11r] and α7 nAChRs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. RgIA and RgIA[11r] Synthesis
4.3. cRNA Preparation and Injection into Xenopus laevis Oocytes
4.4. Electrophysiological Recordings
4.5. Statistical Analysis
4.6. Circular Dichroism (CD) Spectroscopy
4.7. Serum Stability Assay
4.8. Docking
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic acetylcholine receptors: Therapeutic targets for novel ligands to treat pain and inflammation. Pharmacol. Res. 2023, 190, 106715. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martinez, I.E.; Rodriguez, M.C.; Cerbon, M.; Ramos-Martinez, J.C.; Ramos-Martinez, E.G. Role of the Cholinergic Anti-Inflammatory Reflex in Central Nervous System Diseases. Int. J. Mol. Sci. 2021, 22, 13427. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef]
- Patel, R.; Montagut-Bordas, C.; Dickenson, A.H. Calcium channel modulation as a target in chronic pain control. Br. J. Pharmacol. 2018, 175, 2173–2184. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Vetter, I.; Lewis, R.J. Therapeutic potential of cone snail venom peptides (conopeptides). Curr. Top. Med. Chem. 2012, 12, 1546–1552. [Google Scholar] [CrossRef]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Halai, R.; Craik, D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009, 26, 526–536. [Google Scholar] [CrossRef]
- Ellison, M.; Haberlandt, C.; Gomez-Casati, M.E.; Watkins, M.; Elgoyhen, A.B.; McIntosh, J.M.; Olivera, B.M. α-RgIA: A novel conotoxin that specifically and potently blocks the α9α10 nAChR. Biochemistry 2006, 45, 1511–1517. [Google Scholar] [CrossRef]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef]
- Safavi-Hemami, H.; Brogan, S.E.; Olivera, B.M. Pain therapeutics from cone snail venoms: From Ziconotide to novel non-opioid pathways. J. Proteomics 2019, 190, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhu, X.; Xu, P.; Li, R.; Fu, Y.; Dong, S.; Zhangsun, D.; Wu, Y.; Luo, S. d-Amino Acid Substitution of α-Conotoxin RgIA Identifies its Critical Residues and Improves the Enzymatic Stability. Mar. Drugs 2019, 17, 142. [Google Scholar] [CrossRef] [PubMed]
- Zouridakis, M.; Papakyriakou, A.; Ivanov, I.A.; Kasheverov, I.E.; Tsetlin, V.; Tzartos, S.; Giastas, P. Crystal Structure of the Monomeric Extracellular Domain of α9 Nicotinic Receptor Subunit in Complex With α-Conotoxin RgIA: Molecular Dynamics Insights Into RgIA Binding to α9α10 Nicotinic Receptors. Front. Pharm. 2019, 10, 474. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The cholinergic anti-inflammatory pathway: A missing link in neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Baez-Pagan, C.A.; Delgado-Velez, M.; Lasalde-Dominicci, J.A. Activation of the Macrophage α7 Nicotinic Acetylcholine Receptor and Control of Inflammation. J. Neuroimmune Pharmacol. 2015, 10, 468–476. [Google Scholar] [CrossRef]
- Ellison, M.; Gao, F.; Wang, H.L.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. A-conotoxins ImI and ImII target distinct regions of the human α7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. A-selenoconotoxins, a new class of potent α7 neuronal nicotinic receptor antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, X.; Zhangsun, M.; Wu, Y.; Yu, J.; Harvey, P.J.; Kaas, Q.; Zhangsun, D.; Craik, D.J.; Luo, S. Engineered Conotoxin Differentially Blocks and Discriminates Rat and Human α7 Nicotinic Acetylcholine Receptors. J. Med. Chem. 2021, 64, 5620–5631. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, X.; Zhang, L.; Kudryavtsev, D.; Kasheverov, I.; Lei, Y.; Zhangsun, D.; Tsetlin, V.; Luo, S. Species specificity of rat and human α7 nicotinic acetylcholine receptors towards different classes of peptide and protein antagonists. Neuropharmacology 2018, 139, 226–237. [Google Scholar] [CrossRef]
- Kompella, S.N.; Cuny, H.; Hung, A.; Adams, D.J. Molecular Basis for Differential Sensitivity of α-Conotoxin RegIIA at Rat and Human Neuronal Nicotinic Acetylcholine Receptors. Mol. Pharmacol. 2015, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Talley, T.T.; Olivera, B.M.; Han, K.H.; Christensen, S.B.; Dowell, C.; Tsigelny, I.; Ho, K.Y.; Taylor, P.; McIntosh, J.M. A-conotoxin OmIA is a potent ligand for the acetylcholine-binding protein as well as α3beta2 and α7 nicotinic acetylcholine receptors. J. Biol. Chem. 2006, 281, 24678–24686. [Google Scholar] [CrossRef]
- Armishaw, C.; Jensen, A.A.; Balle, T.; Clark, R.J.; Harpsoe, K.; Skonberg, C.; Liljefors, T.; Stromgaard, K. Rational design of α-conotoxin analogues targeting α7 nicotinic acetylcholine receptors: Improved antagonistic activity by incorporation of proline derivatives. J. Biol. Chem. 2009, 284, 9498–9512. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, P.; Christensen, S.; Yoshikami, D.; Dowell, C.; Watkins, M.; Gulyas, J.; Rivier, J.; Olivera, B.M.; McIntosh, J.M. Discovery, synthesis, and structure activity of a highly selective α7 nicotinic acetylcholine receptor antagonist. Biochemistry 2007, 46, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Inserra, M.C.; Kompella, S.N.; Vetter, I.; Brust, A.; Daly, N.L.; Cuny, H.; Craik, D.J.; Alewood, P.F.; Adams, D.J.; Lewis, R.J. Isolation and characterization of α-conotoxin LsIA with potent activity at nicotinic acetylcholine receptors. Biochem. Pharmacol. 2013, 86, 791–799. [Google Scholar] [CrossRef]
- Nicke, A.; Samochocki, M.; Loughnan, M.L.; Bansal, P.S.; Maelicke, A.; Lewis, R.J. A-conotoxins EpI and AuIB switch subtype selectivity and activity in native versus recombinant nicotinic acetylcholine receptors. FEBS Lett. 2003, 554, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.N.T.; Lee, H.S.; Swaminathan, S.; Goodwin, L.; Rai, N.; Ushay, B.; Lewis, R.J.; Rosengren, K.J.; Conibear, A.C. Posttranslational modifications of α-conotoxins: Sulfotyrosine and C-terminal amidation stabilise structures and increase acetylcholine receptor binding. RSC. Med. Chem. 2021, 12, 1574–1584. [Google Scholar] [CrossRef]
- Millard, E.L.; Nevin, S.T.; Loughnan, M.L.; Nicke, A.; Clark, R.J.; Alewood, P.F.; Lewis, R.J.; Adams, D.J.; Craik, D.J.; Daly, N.L. Inhibition of Neuronal Nicotinic Acetylcholine Receptor Subtypes by α-Conotoxin GID and Analogues. J. Biol. Chem. 2009, 284, 4944–4951. [Google Scholar] [CrossRef]
- Nicke, A.; Loughnan, M.L.; Millard, E.L.; Alewood, P.F.; Adams, D.J.; Daly, N.L.; Craik, D.J.; Lewis, R.J. Isolation, structure, and activity of GID, a novel α 4/7-conotoxin with an extended N-terminal sequence. J. Biol. Chem. 2003, 278, 3137–3144. [Google Scholar] [CrossRef]
- Tugyi, R.; Uray, K.; Ivan, D.; Fellinger, E.; Perkins, A.; Hudecz, F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar] [CrossRef]
- Shen, J. D-Amino acid substituted peptides as potential alternatives of homochiral L-configurations. Amino Acids 2021, 53, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Zhu, X.; Wu, Y.; Hu, Y.; Christensen, S.; Harvey, P.J.; Akcan, M.; Craik, D.J.; McIntosh, J.M. Characterization of a novel α-conotoxin TxID from Conus textile that potently blocks rat α3beta4 nicotinic acetylcholine receptors. J. Med. Chem. 2013, 56, 9655–9663. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Schroeder, C.I.; Zhu, X.; Hu, Y.; Wu, Y.; Weltzin, M.M.; Eberhard, S.; Kaas, Q.; Craik, D.J.; et al. A novel α4/7-conotoxin LvIA from Conus lividus that selectively blocks α3beta2 vs. α6/α3beta2beta3 nicotinic acetylcholine receptors. FASEB J. 2014, 28, 1842–1853. [Google Scholar] [CrossRef]
- White, A.M.; de Veer, S.J.; Wu, G.; Harvey, P.J.; Yap, K.; King, G.J.; Swedberg, J.E.; Wang, C.K.; Law, R.H.P.; Durek, T.; et al. Application and structural analysis of triazole-bridged disulfide mimetics in cyclic peptides. Angew. Chem. 2020, 59, 11273–11277. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptors | RgIA | RgIA[11r] | IC50 Ratio of RgIA/RgIA[11r] | ||||
|---|---|---|---|---|---|---|---|
| IC50 (95% CI *) (nM) | Hill Slope | n | IC50 (95% CI *) (nM) | Hill Slope | n | ||
| rα7 | 5313 (3068–9202) | 0.8 (0.4–1.1) | 3 | 163 (143–187) | 0.8 (0.7–0.9) | 11 | 32.6 |
| hα7 | 4608 (2516–8440) | 0.7 (0.4–1.0) | 4 | 463 (333–640) | 0.6 (0.5–0.7) | 7 | 9.96 |
| rα9α10 | 2.6 (2.1–3.2) | 0.8 (0.7–0.9) | 11 | 15,820 (10,380–24,200) | 0.8 (0.5–1.2) | 6 | 0.00016 |
| hα9α10 | 1398 (997–1961) | 0.5 (0.4–0.6) | 6 | >10,000 | - | 3 | - |
| Name | Sequences | α7 (IC50, nM) | Other nAChR Subtypes | Ref. |
|---|---|---|---|---|
| RgIA[11r] | GCCSDPRCRYrCR | rα7 (163) > hα7 (463) | >10,000 | This work |
| RegIIA | GCCSHPACNVNNPHIC # | rα7 (41) > hα7 (210) | α3β2 > α3β4 > α7 α6/α3β4β3 | [20,21] |
| [H5D]RegIIA | GCCSDPACNVNNPHIC # | rα7 (100) > hα7 (13,680) | N.D. | [20] |
| OmIA | GCCSHPACNVNNPHICG # | rα7 (59) > hα7 (290) | α3β2 > αα6/α3β2 > α7 | [22] |
| PnIA | GCCSLPPCAANNPD(sTy)C # | hα7-5HT3 chimaera (510), rα7 (252) | α3β2 > α7 | [23,24] |
| PnIA [A10L,sTy15Y] | GCCSLPPCALNNPDYC # | rα7 (12) > hα7 (59) | α3β2 ≈ α7 | [20] |
| [Q1G,ΔR14]LvIB | GCCSNPPCAHEHC # | rα7 (97) > hα7 (1570) | α7 > α6/α3β2β3 > rα3β2 > rα6/α3β4 | [19] |
| ImI | GCCSDPRCAWRC # | rα7 (69) > hα7 (440) | hα3β2 ≈ α7 | [17,18] |
| ImII | ACCSDPRCAWRC # | rα7 (441) > hα7 (571) | α7 > α1β1δε | [17] |
| LsIA | SGCCSNPACRVNNPNIC # | hα7 (10.1) | α7 ≈ α3β2 > α3α5β2 | [25] |
| ArIA | IRDECCSNPACRVNNPHVCRRR | rα7 (6.02) | α7 >α3β2 | [24] |
| ArIB | DECCSNPACRVNNPHVCRRR | rα7 (1.81) | α7 > α6/α3β2β3 > α3β2 | [24] |
| ArIB [V11L, V16A] | DECCSNPACRLNNPHACRRR | rα7 (0.356) | α7 > α6/α3β2β3 > α3β2 | [24] |
| EpI | GCCSDPRCNMNNPD(sTy)C # | rα7 (30) | α7 > α3β4 | [26,27] |
| GID | IRD(Gla)CCSNPACRVNNOHVC | hα7 (4.5) > rα7 (5.1) | α3β2 > α7 > α3β4 | [28,29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zhang, J.; Ren, J.; Zhu, X.; Li, R.; Zhangsun, D.; Luo, S. Substitution of D-Arginine at Position 11 of α-RgIA Potently Inhibits α7 Nicotinic Acetylcholine Receptor. Mar. Drugs 2023, 21, 326. https://doi.org/10.3390/md21060326
Wu Y, Zhang J, Ren J, Zhu X, Li R, Zhangsun D, Luo S. Substitution of D-Arginine at Position 11 of α-RgIA Potently Inhibits α7 Nicotinic Acetylcholine Receptor. Marine Drugs. 2023; 21(6):326. https://doi.org/10.3390/md21060326
Chicago/Turabian StyleWu, Yong, Junjie Zhang, Jie Ren, Xiaopeng Zhu, Rui Li, Dongting Zhangsun, and Sulan Luo. 2023. "Substitution of D-Arginine at Position 11 of α-RgIA Potently Inhibits α7 Nicotinic Acetylcholine Receptor" Marine Drugs 21, no. 6: 326. https://doi.org/10.3390/md21060326
APA StyleWu, Y., Zhang, J., Ren, J., Zhu, X., Li, R., Zhangsun, D., & Luo, S. (2023). Substitution of D-Arginine at Position 11 of α-RgIA Potently Inhibits α7 Nicotinic Acetylcholine Receptor. Marine Drugs, 21(6), 326. https://doi.org/10.3390/md21060326