
Jejucarbosides B–E, Chlorinated Cycloaromatized Enediynes, from a Marine Streptomyces sp.


Abstract
:
1. Introduction
2. Results and Discussion
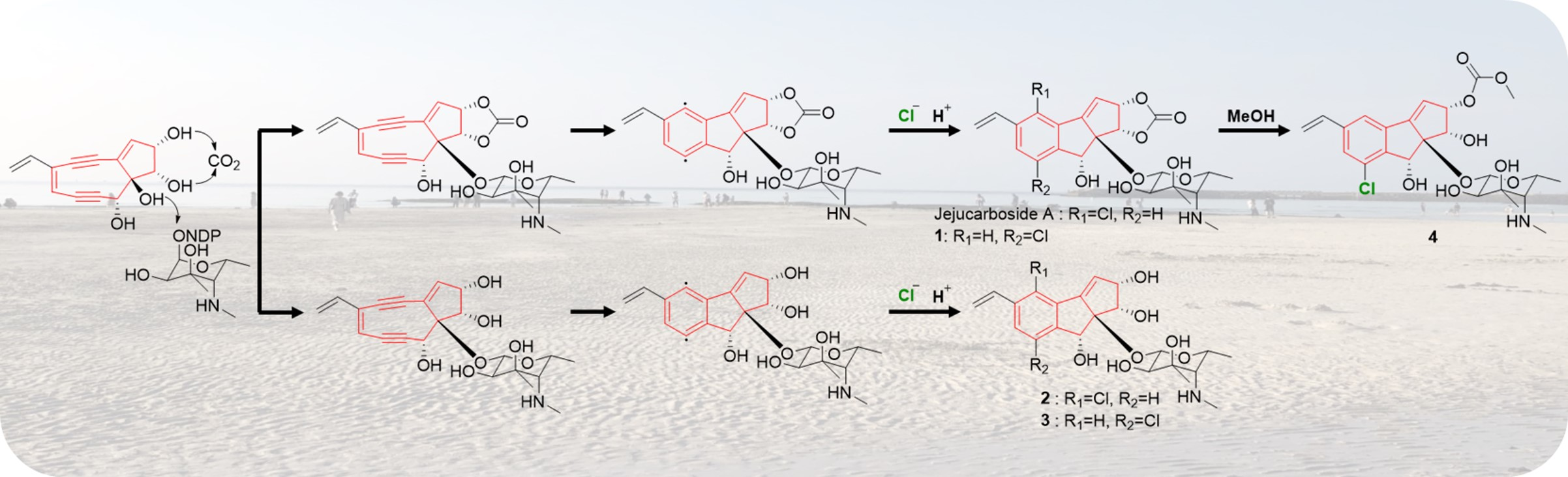
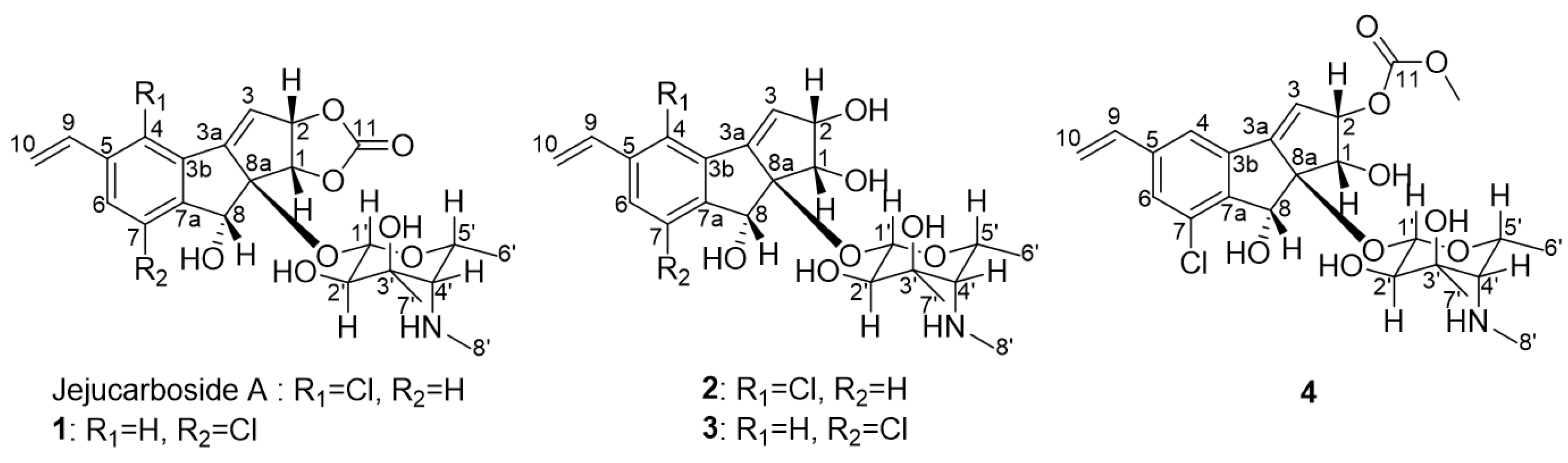
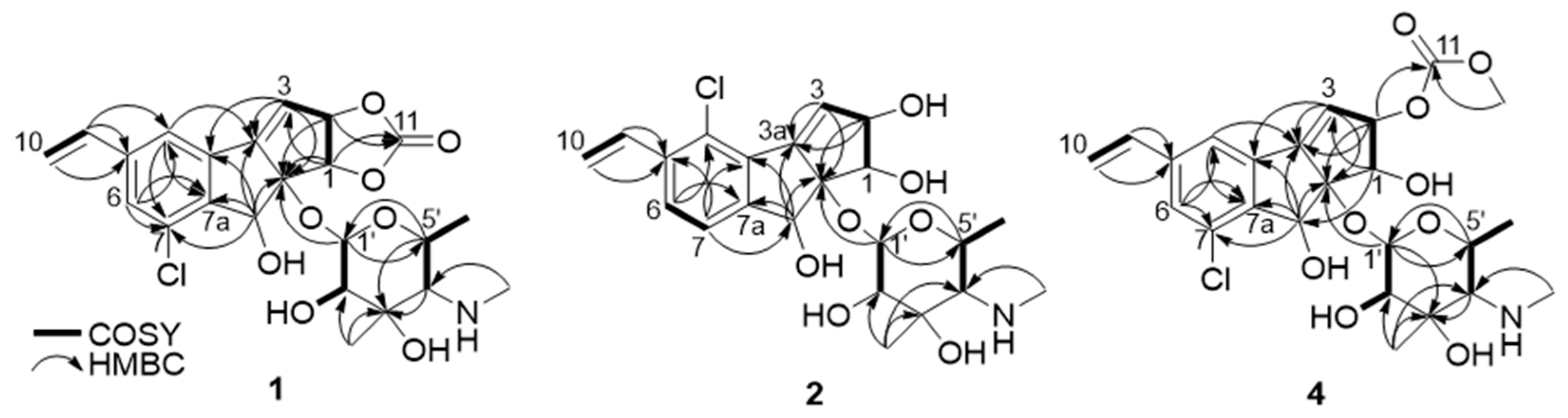
2.1. Discovery and Structure Elucidation
2.2. Proposed Mechanism of Production of Jejucarbosides B–E
2.3. Biological Activity Jejucarbosides B–E
3. Materials and Methods
3.1. General Experiment Procedures
3.2. Bacterial Strain and Subculture Selection
3.3. Scale-up Culture and Extraction
3.4. Isolation of Jejucarbosides B–E (1–4)
3.5. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Shen, B.; Hindra; Yan, X.; Huang, T.; Ge, H.; Yang, D.; Teng, Q.; Rudolf, J.D.; Lohman, J.R. Enediynes: Exploration of Microbial Genomics to Discover New Anticancer Drug Leads. Bioorg. Med. Chem. Lett. 2015, 25, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Perrin, C.L.; Rodgers, B.L.; O’Connor, J.M. Nucleophilic Addition to a p-Benzyne Derived from an Enediyne: A New Mechanism for Halide Incorporation into Biomolecules. J. Am. Chem. Soc. 2007, 129, 4795–4799. [Google Scholar] [CrossRef]
- Liang, Z.-X. Complexity and Simplicity in the Biosynthesis of Enediyne Natural Products. Nat. Prod. Rep. 2010, 27, 499–528. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Kumagai, K.; Ishida, N. Characterization of Neocarzinostatin. J. Antibiot. 1996, 19, 253–259. [Google Scholar]
- Maeda, H.; Edo, K.; Ishida, N. Neocarzinostatin: The Past, Present, and Future of an Anticancer Drug; Springer: New York, NY, USA, 1997. [Google Scholar]
- Otani, T.; Minami, Y.; Marunaka, T. A New Macromolecular Antitumor Antibiotic, C-1027 II. Isolation and Physico-Chemical Properties. J. Antibiot. 1988, 41, 1580–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Xue, Y.-C.; Xie, M.-Y.; Zhang, R.; Minami, Y.; Yamada, Y.; Marunaka, T. A New Macromolecular Antitumor Antibiotic, C-1027. I. Discovery, Taxonomy of Producing Organism, Fermentation and Biological Activity. J. Antibiot. 1988, 41, 1575–1579. [Google Scholar] [CrossRef] [Green Version]
- Lam, K.S.; Hesler, G.A.; Gustavson, D.R.; Crosswell, A.R.; Veitch, J.M.; Forenza, S. Kedarcidin, A New Chromoprotein Antitumor Antibiotic. I. Taxonomy of Producing Organism, Fermentation and Biological Activity. J. Antibiot. 1991, 44, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Hanada, M.; Ohkuma, H.; Yonemoto, T.; Tomita, K.; Ohbayashi, M.; Kamei, H.; Miyaki, T.; Konishi, M.; Kawaguchi, H. Maduropeptin, A Complex of New Macromolecular Antitumor Antibiotics. J. Antibiot. 1991, 44, 403–414. [Google Scholar] [CrossRef]
- Ando, T.; Ishii, M.; Kajiura, T.; Kameyama, T.; Miwa, K.; Sugiura, Y. A New Non-Protein Enediyne Antibiotic N1999A2: Unique Enediyne Chromophore Similar to Neocarzinostatin and DNA Cleavage Feature. Tetrahedron Lett. 1998, 39, 6495–6498. [Google Scholar] [CrossRef]
- Buchanan, G.O.; Williams, P.G.; Feling, R.H.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Sporolides A and B: Structurally Unprecedented Halogenated Macrolides from the Marine Actinomycete Salinispora tropica. Org. Lett. 2005, 7, 2731–2734. [Google Scholar] [CrossRef]
- Oh, D.-C.; Williams, P.G.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Cyanosporasides A and B, Chloro- and Cyano-cyclopenta[a]indene Glycosides from the Marine Actinomycete “Salinispora pacifica”. Org. Lett. 2006, 8, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.-J.; Gaudencio, S.P.; Kauffman, C.A.; Jensen, P.R.; Kondratyuk, T.P.; Marler, L.E.; Pezzuto, J.M.; Fenical, W. Fijiolides A and B, Inhibitors of TNF-α-induced NFκB Activation, from a Marine-Derived Sediment Bacterium of the Genus Nocardiopsis. J. Nat. Prod. 2010, 73, 1080–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.Y.; Xiao, Y.S.; Zhang, B.; Shao, F.L.; Guo, Z.K.; Zhang, J.J.; Jiao, R.H.; Sun, Y.; Xu, Q.; Tan, R.X.; et al. Amycolamycins A and B, Two Enediyne-Derived Compounds from a Locust-Associated Actinomycete. Org. Lett. 2017, 19, 6208–6211. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tan, Q.; Zhu, S.; Yan, X.; Li, Y.; Zhuang, Z.; Zhu, X.; Duan, Y.; Huang, Y. Discovery of Pentaene Polyols by the Activation of an Enediyne Gene Clusters: Biosynthetic Implications for 9-memebere Enediyne Core Structures. Chem. Sci. 2022, 13, 13475–13481. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Cui, Z.; Hankore, E.D.; Moonschi, F.H.; Esfahani, H.S.; Kalkreuter, E.; Gui, C.; Yang, D.; Phillips, G.N.; Thorson, J.S.; et al. A Discrete Intermediate for the Biosynthesis of Both the Enediyne Core and the Anthraquinone Moiety of Enediyne Natural Products. Proc. Natl. Acad. Sci. USA 2023, 120, e2220468120. [Google Scholar] [CrossRef] [PubMed]
- Im, J.H.; Shin, D.; Ban, Y.H.; Byun, W.S.; Bae, E.S.; Lee, D.; Du, Y.E.; Cui, J.; Kwon, Y.; Nam, S.-J.; et al. Targeted Discovery of an Enediyne-Derived Cycloaromatized Compound, Jejucarboside A, from a Marine Actinomycete. Org. Lett. 2022, 24, 7188–7193. [Google Scholar] [CrossRef]
- Pretsch, E.; Bűhlmann, P.; Affolter, C. Structure Determination of Organic Compounds-Tables of Spectral Data; Springer: New York, NY, USA, 2000; p. 153. [Google Scholar]
- Byun, W.S.; Kim, S.; Shin, Y.H.; Kim, W.K.; Oh, D.-C.; Lee, S.K. Antitumor Activity of Ohmyungsamycin A through the Regulation of the Skp2-p27 Axis and MCM4 in Human Colorectal Cancer Cells. J. Nat. Prod. 2020, 83, 118–126. [Google Scholar] [CrossRef]
- Yan, X.; Sims, J.; Wang, B.; Hamann, M.T. Marine Actinomycete Streptomyces sp. ISP2-42E, A New Source of Rhamnolipid. Biochem. Syst. Ecol. 2014, 55, 292–295. [Google Scholar] [CrossRef] [Green Version]
- Vichai, V.; Kirtikara, K. Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Jejucarboside B (1) | Jejucarboside C (2) | Jejucarboside D (3) | Jejucarboside E (4) | |||||
|---|---|---|---|---|---|---|---|---|
| Position | δC, Type | δH, Mult (J in Hz) | δC, Type | δH, Mult (J in Hz) | δC, Type | δH, Mult (J in Hz) | δC, Type | δH, Mult (J in Hz) |
| 1 | 74.6, CH | 5.34, d (7.0) | 70.5, CH | 4.25, d (5.5) | 70.7, CH | 4.28, d (5.0) | 68.9, CH | 4.54, d (6.0) |
| 2 | 87.5, CH | 5.57, dd (7.0, 3.0) | 79.8, CH | 4.59, dd (5.5, 3.0) | 79.6, CH | 4.58, dd (5.0, 3.0) | 84.0, CH | 5.61, dd (6.0, 3.0) |
| 3 | 122.4, CH | 6.30, d (3.0) | 129.9, CH | 6.57. d (3.0) | 127.6, CH | 6.34, d (3.0) | 123.0, CH | 6.27, d (3.0) |
| 3a | 156.5, C | 150.3, C | 152.0, C | 154.6, C | ||||
| 3b | 137.7, C | 134.7, C | 138.4, C | 137.8, C | ||||
| 4 | 120.3, CH | 7.68, s | 129.6, C | 120.1, CH | 7.58, s | 120.5, CH | 7.60, s | |
| 5 | 142.1, C | 137.5, C | 141.8, C | 141.8, C | ||||
| 6 | 129.2, CH | 7.49, s | 128.8, CH | 7.63, d (8.0) | 128.3, CH | 7.42, s | 128.8, CH | 7.44, s |
| 7 | 133.2, C | 126.5, CH | 7.40, d (8.0) | 133.5, C | 133.5, C | |||
| 7a | 146.6, C | 152.6, C | 147.4, C | 147.6, C | ||||
| 8 | 73.3, CH | 5.21, s | 73.8, CH | 5.01, s | 72.5, CH | 5.19, s | 72.7, CH | 5.13, s |
| 8a | 95.1, C | 96.6, C | 96.2, C | 96.4, C | ||||
| 9 | 135.9, CH | 6.76, dd (18.0, 11.0) | 133.2, CH | 7.13, dd (17.5, 11.0) | 136.2, CH | 6.75, dd (18.0, 11.0) | 136.1, CH | 6.73, dd (17.5, 11.0) |
| 10 | 117.4, CH2 | 5.40, d (11.0) | 118.3, CH2 | 5.46, d (11.0) | 116.9, CH2 | 5.36, d (11.0) | 117.1, CH2 | 5.36, d (11.0) |
| 5.92, d (18.0) | 5.85, d (17.5) | 5.88, d (18.0) | 5.88, d (17.5) | |||||
| 11 | 156.5, C | 156.2, C | ||||||
| 11-OMe | 55.5, CH3 | 3.77, s | ||||||
| 1′ | 99.7, CH | 4.38, d (7.5) | 98.6, CH | 4.95, d (8.0) | 99.3, CH | 5.02, d (8.0) | 98.3, CH | 4.93, d (8.0) |
| 2′ | 72.8, CH | 2.99, d (7.5) | 73.3, CH | 3.08, d (8.0) | 72.8, CH | 3.09, d (8.0) | 73.1, CH | 3.07, d (8.0) |
| 3′ | 76.0, C | 76.1, C | 75.9, C | 76.1, C | ||||
| 4′ | 68.8, CH | 1.88, s | 68.8, CH | 1.95, d (1.5) | 68.8, CH | 1.97, d (1.5) | 68.8, CH | 2.00, d (1.5) |
| 5′ | 70.0, CH | 3.97, q (6.5) | 70.3, CH | 4.20, qd (6.5, 1.5) | 70.1, CH | 4.24, qd (6.5, 1.5) | 70.4, CH | 4.32, qd (6.5, 1.5) |
| 6′ | 16.6, CH3 | 1.10, d (6.5) | 17.0, CH3 | 1.13, d (6.5) | 16.5, CH3 | 1.15, d (6.5) | 17.0, CH3 | 1.17, d (6.5) |
| 7′ | 23.6, CH3 | 1.19, s | 23.6, CH3 | 1.22, s | 23.7, CH3 | 1.23, s | 23.6, CH3 | 1.23, s |
| 8′ | 39.1, CH3 | 2.40, s | 39.1, CH3 | 2.41, s | 39.1, CH3 | 2.41, s | 39.1, CH3 | 2.43, s |
| (μM) | SNU638 | SK-Hep-1 | A549 | HCT116 | MDA-MB-231 |
|---|---|---|---|---|---|
| Jejucarboside A | >100 | >100 | >100 | 29.30 | >100 |
| Jejucarboside B | 14.17 | 41.55 | 26.09 | 16.47 | 42.38 |
| Jejucarboside C | 16.40 | 46.63 | 25.17 | 21.33 | 43.20 |
| Jejucarboside D | >100 | >100 | >100 | >100 | >100 |
| Jejucarboside E | 0.31 | 0.40 | 0.25 | 0.29 | 0.48 |
| Etoposide | 0.20 | 0.63 | 0.21 | 0.56 | 4.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Im, J.H.; Shin, Y.-H.; Bae, E.S.; Lee, S.K.; Oh, D.-C. Jejucarbosides B–E, Chlorinated Cycloaromatized Enediynes, from a Marine Streptomyces sp. Mar. Drugs 2023, 21, 405. https://doi.org/10.3390/md21070405
Im JH, Shin Y-H, Bae ES, Lee SK, Oh D-C. Jejucarbosides B–E, Chlorinated Cycloaromatized Enediynes, from a Marine Streptomyces sp. Marine Drugs. 2023; 21(7):405. https://doi.org/10.3390/md21070405
Chicago/Turabian StyleIm, Ji Hyeon, Yern-Hyerk Shin, Eun Seo Bae, Sang Kook Lee, and Dong-Chan Oh. 2023. "Jejucarbosides B–E, Chlorinated Cycloaromatized Enediynes, from a Marine Streptomyces sp." Marine Drugs 21, no. 7: 405. https://doi.org/10.3390/md21070405
APA StyleIm, J. H., Shin, Y. -H., Bae, E. S., Lee, S. K., & Oh, D. -C. (2023). Jejucarbosides B–E, Chlorinated Cycloaromatized Enediynes, from a Marine Streptomyces sp. Marine Drugs, 21(7), 405. https://doi.org/10.3390/md21070405