

An Investigation of Structure–Activity Relationships and Cell Death Mechanisms of the Marine Alkaloids Discorhabdins in Merkel Cell Carcinoma Cells

 , , , , ,
, , , , ,
Abstract
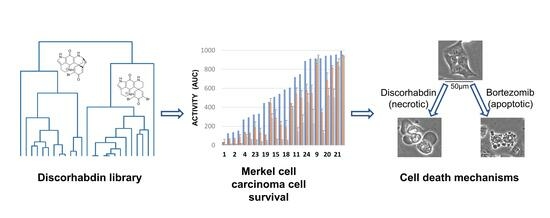
:
1. Introduction
2. Results
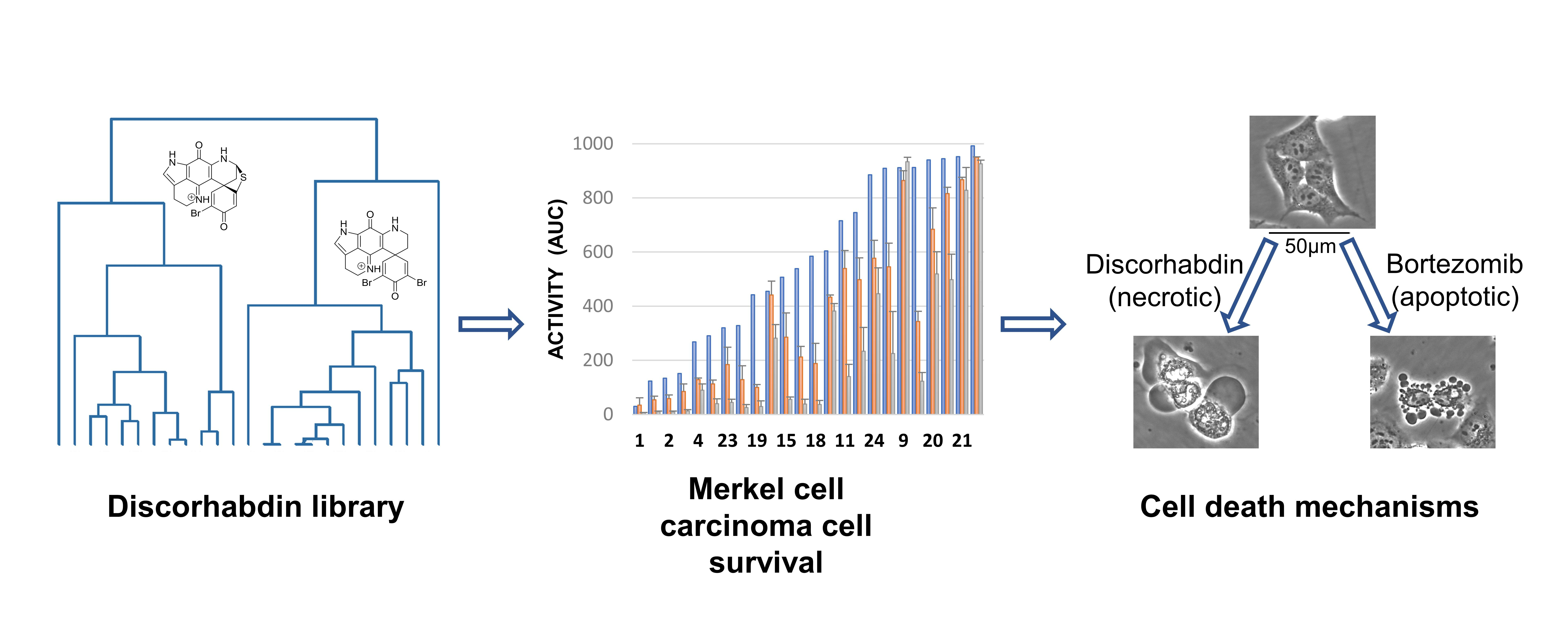
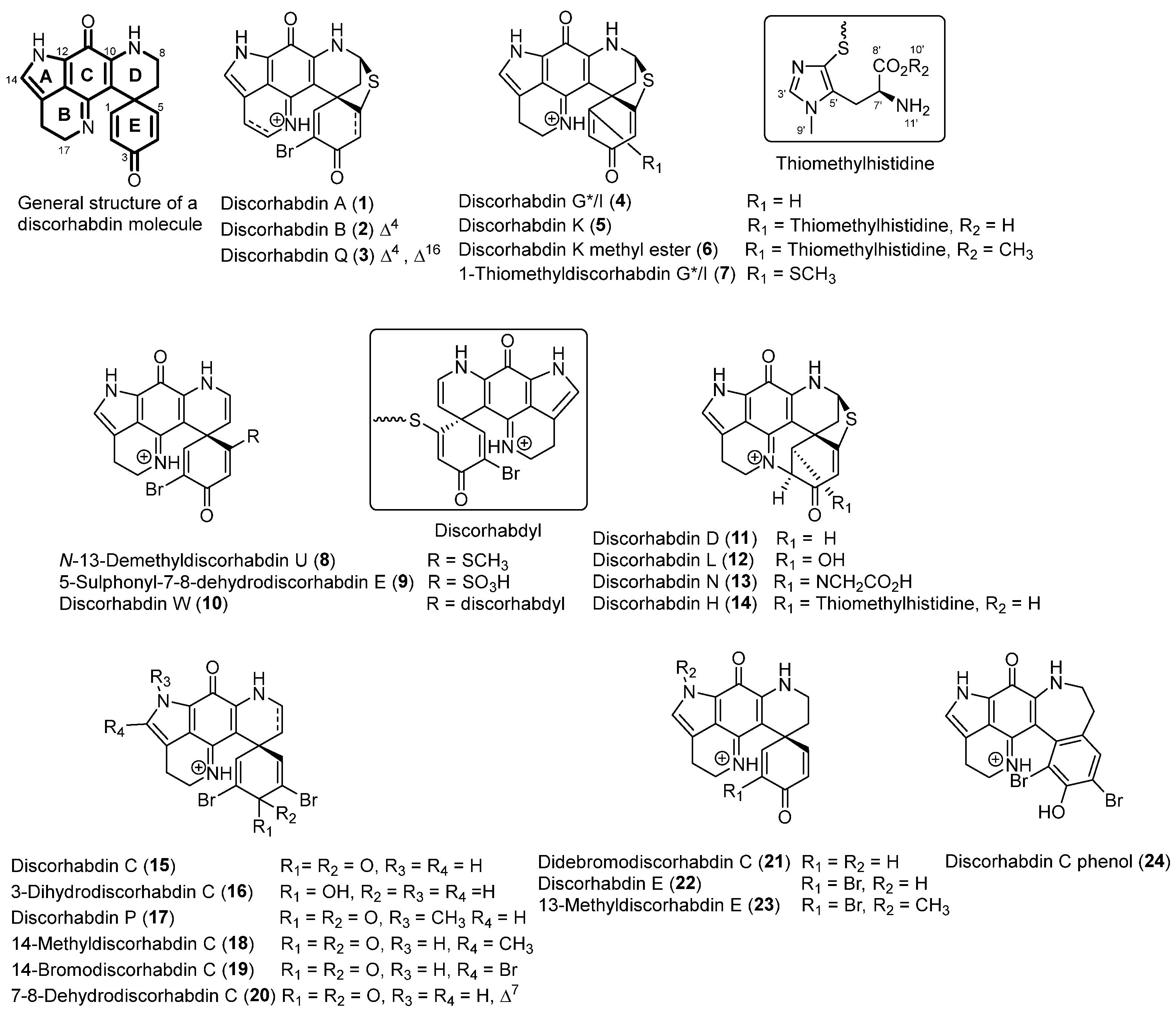
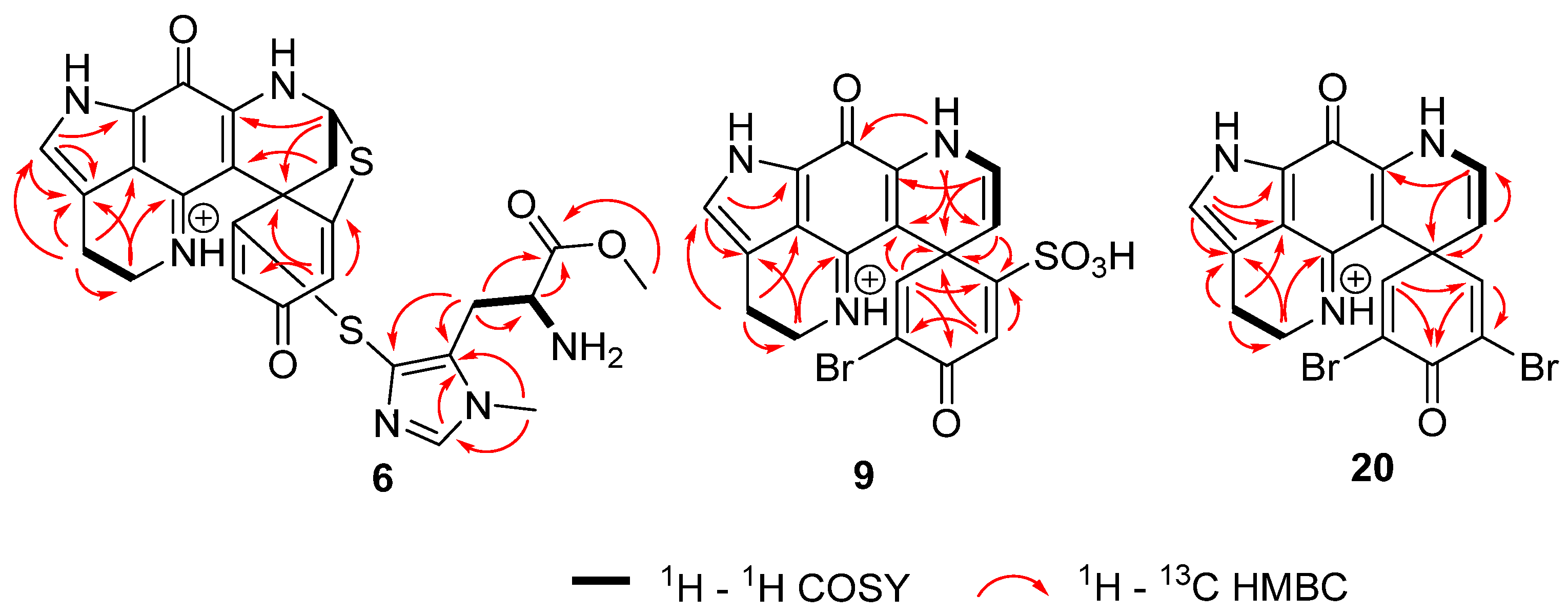
2.1. Structure Elucidation
2.2. Assay Activity
2.3. Cell Death Mechanisms
2.4. Mitochondrial Dysfunction
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Chemicals and Reagents
4.3. Natural Product Isolation
4.4. Preparation of Semi-Synthetic Derivatives
4.5. Compound Characterization
4.6. Computational Methods
4.7. Cell lines Used
4.8. Cell Viability and Analysis of Cell Death Mechanisms
4.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef]
- Patel, P.; Hussain, K. Merkel cell carcinoma. Clin. Exp. Dermatol. 2021, 46, 814–819. [Google Scholar] [CrossRef]
- Zwijnenburg, E.M.; Lubeek, S.F.K.; Werner, J.E.M.; Amir, A.L.; Weijs, W.L.J.; Takes, R.P.; Pegge, S.A.H.; van Herpen, C.M.L.; Adema, G.J.; Kaanders, J. Merkel Cell Carcinoma: New Trends. Cancers 2021, 13, 1614. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.M.; Cerroni, L. Merkel cell carcinoma: A review. J. Cutan. Pathol. 2021, 48, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Banks, P.D.; Sandhu, S.; Gyorki, D.E.; Johnston, M.L.; Rischin, D. Recent Insights and Advances in the Management of Merkel Cell Carcinoma. J. Oncol. Pract. 2016, 12, 637–646. [Google Scholar] [CrossRef]
- Angeles, C.V.; Sabel, M.S. Immunotherapy for Merkel cell carcinoma. J. Surg. Oncol. 2021, 123, 775–781. [Google Scholar] [CrossRef]
- Harms, K.L.; Zhao, L.; Johnson, B.; Wang, X.; Carskadon, S.; Palanisamy, N.; Rhodes, D.R.; Mannan, R.; Vo, J.N.; Choi, J.E.; et al. Virus-positive Merkel Cell Carcinoma Is an Independent Prognostic Group with Distinct Predictive Biomarkers. Clin. Cancer Res. 2021, 27, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Samimi, M. Immune Checkpoint Inhibitors and Beyond: An Overview of Immune-Based Therapies in Merkel Cell Carcinoma. Am. J. Clin. Dermatol. 2019, 20, 391–407. [Google Scholar] [CrossRef]
- Iyer, J.G.; Blom, A.; Doumani, R.; Lewis, C.; Tarabadkar, E.S.; Anderson, A.; Ma, C.; Bestick, A.; Parvathaneni, U.; Bhatia, S.; et al. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 2016, 5, 2294–2301. [Google Scholar] [CrossRef]
- Hill, N.T.; Kim, D.; Busam, K.J.; Chu, E.Y.; Green, C.; Brownell, I. Distinct Signatures of Genomic Copy Number Variants Define Subgroups of Merkel Cell Carcinoma Tumors. Cancers 2021, 13, 1134. [Google Scholar] [CrossRef]
- Shao, Q.; Byrum, S.D.; Moreland, L.E.; Mackintosh, S.G.; Kannan, A.; Lin, Z.; Morgan, M.; Stack, B.C., Jr.; Cornelius, L.A.; Tackett, A.J.; et al. A Proteomic Study of Human Merkel Cell Carcinoma. J. Proteom. Bioinform. 2013, 6, 275–282. [Google Scholar] [CrossRef]
- Liu, G.Y.; Frank, N.; Bartsch, H.; Lin, J.K. Induction of apoptosis by thiuramdisulfides, the reactive metabolites of dithiocarbamates, through coordinative modulation of NFκB, c-fos/c-jun, and p53 proteins. Mol. Carcinog. 1998, 22, 235–246. [Google Scholar] [CrossRef]
- Schreck, R.; Meier, B.; Männel, D.N.; Dröge, W.; Baeuerle, P.A. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J. Exp. Med. 1992, 175, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Panda, M.; Biswal, B.K. Emerging role of plumbagin: Cytotoxic potential and pharmaceutical relevance towards cancer therapy. Food Chem. Toxicol. 2019, 125, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Binoy, A.; Nedungadi, D.; Katiyar, N.; Bose, C.; Shankarappa, S.A.; Nair, B.G.; Mishra, N. Plumbagin induces paraptosis in cancer cells by disrupting the sulfhydryl homeostasis and proteasomal function. Chem. Biol. Interact. 2019, 310, 108733. [Google Scholar] [CrossRef]
- Blasiak, J. DNA-Damaging Anticancer Drugs—A Perspective for DNA Repair-Oriented Therapy. Curr. Med. Chem. 2017, 24, 1488–1503. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Fukamiya, N.; Lee, K.H.; Muhammad, I.; Murakami, C.; Okano, M.; Harvey, I.; Pelletier, J. Structure-activity relationships of quassinoids for eukaryotic protein synthesis. Cancer Lett. 2005, 220, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhao, S.; Fash, D.M.; Li, Z.; Chain, W.J.; Beutler, J.A. Englerins: A Comprehensive Review. J. Nat. Prod. 2017, 80, 771–781. [Google Scholar] [CrossRef]
- Martirosyan, A.; Leonard, S.; Shi, X.; Griffith, B.; Gannett, P.; Strobl, J. Actions of a histone deacetylase inhibitor NSC3852 (5-nitroso-8-quinolinol) link reactive oxygen species to cell differentiation and apoptosis in MCF-7 human mammary tumor cells. J. Pharmacol. Exp. Ther. 2006, 317, 546–552. [Google Scholar] [CrossRef]
- Smith, E.A.; Hill, N.T.; Gelb, T.; Garman, K.A.; Goncharova, E.I.; Bokesch, H.R.; Kim, C.K.; Wendt, K.L.; Cichewicz, R.H.; Gustafson, K.R.; et al. Identification of natural product modulators of Merkel cell carcinoma cell growth and survival. Sci. Rep. 2021, 11, 13597. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.F.; Fan, H.; Xiong, J.; Wu, S.B. Discorhabdins and pyrroloiminoquinone-related alkaloids. Chem. Rev. 2011, 111, 5465–5491. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Kelly, M.; Tasdemir, D. Chemistry, Chemotaxonomy and Biological Activity of the Latrunculid Sponges (Order Poecilosclerida, Family Latrunculiidae). Mar. Drugs 2021, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Antunes, E.M.; Copp, B.R.; Davies-Coleman, M.T.; Samaai, T. Pyrroloiminoquinone and related metabolites from marine sponges. Nat. Prod. Rep. 2005, 22, 62–72. [Google Scholar] [CrossRef]
- Grkovic, T.; Pearce, A.N.; Munro, M.H.G.; Blunt, J.W.; Davies-Coleman, M.T.; Copp, B.R. Isolation and Characterization of Diastereomers of Discorhabdins H and K and Assignment of Absolute Configuration to Discorhabdins D, N, Q, S, T, and U. J. Nat. Prod. 2010, 73, 1686–1693. [Google Scholar] [CrossRef]
- Lam, C.F.C.; Cadelis, M.M.; Copp, B.R. Exploration of the Electrophilic Reactivity of the Cytotoxic Marine Alkaloid Discorhabdin C and Subsequent Discovery of a New Dimeric C-1/N-13-Linked Discorhabdin Natural Product. Mar. Drugs 2020, 18, 404. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef]
- Song, L.; Bretz, A.C.; Gravemeyer, J.; Spassova, I.; Muminova, S.; Gambichler, T.; Sriram, A.; Ferrone, S.; Becker, J.C. The HDAC Inhibitor Domatinostat Promotes Cell-Cycle Arrest, Induces Apoptosis, and Increases Immunogenicity of Merkel Cell Carcinoma Cells. J. Investig. Dermatol. 2021, 141, 903–912.e4. [Google Scholar] [CrossRef]
- Fang, B.; Kannan, A.; Zhao, S.; Nguyen, Q.H.; Ejadi, S.; Yamamoto, M.; Camilo Barreto, J.; Zhao, H.; Gao, L. Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts. Sci. Rep. 2020, 10, 8867. [Google Scholar] [CrossRef]
- Adam, C.; Baeurle, A.; Brodsky, J.L.; Wipf, P.; Schrama, D.; Becker, J.C.; Houben, R. The HSP70 modulator MAL3-101 inhibits Merkel cell carcinoma. PLoS ONE 2014, 9, e92041. [Google Scholar] [CrossRef] [PubMed]
- Das, B.K.; Kannan, A.; Nguyen, Q.; Gogoi, J.; Zhao, H.; Gao, L. Selective Inhibition of Aurora Kinase A by AK-01/LY3295668 Attenuates MCC Tumor Growth by Inducing MCC Cell Cycle Arrest and Apoptosis. Cancers 2021, 13, 3708. [Google Scholar] [CrossRef] [PubMed]
- Kadletz, L.; Bigenzahn, J.; Thurnher, D.; Stanisz, I.; Erovic, B.M.; Schneider, S.; Schmid, R.; Seemann, R.; Birner, P.; Heiduschka, G. Evaluation of Polo-like kinase 1 as a potential therapeutic target in Merkel cell carcinoma. Head Neck 2016, 38 (Suppl. S1), E1918–E1925. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.E.; Mangelberger, D.; Weick, J.W.; Vozheiko, T.D.; Harms, P.W.; Nash, K.T.; Quintana, E.; Baciu, P.; Johnson, T.M.; Bichakjian, C.K.; et al. Merkel cell carcinoma dependence on bcl-2 family members for survival. J. Investig. Dermatol. 2014, 134, 2241–2250. [Google Scholar] [CrossRef]
- Kannan, A.; Lin, Z.; Shao, Q.; Zhao, S.; Fang, B.; Moreno, M.A.; Vural, E.; Stack, B.C., Jr.; Suen, J.Y.; Kannan, K.; et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget 2016, 7, 6576–6592. [Google Scholar] [CrossRef]
- Leiendecker, L.; Jung, P.S.; Krecioch, I.; Neumann, T.; Schleiffer, A.; Mechtler, K.; Wiesner, T.; Obenauf, A.C. LSD1 inhibition induces differentiation and cell death in Merkel cell carcinoma. EMBO Mol. Med. 2020, 12, e12525. [Google Scholar] [CrossRef]
- Sarma, B.; Willmes, C.; Angerer, L.; Adam, C.; Becker, J.C.; Kervarrec, T.; Schrama, D.; Houben, R. Artesunate Affects T Antigen Expression and Survival of Virus-Positive Merkel Cell Carcinoma. Cancers 2020, 12, 919. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; McDermott, A.; Shao, L.; Kannan, A.; Morgan, M.; Stack, B.C., Jr.; Moreno, M.; Davis, D.A.; Cornelius, L.A.; Gao, L. Chronic mTOR activation promotes cell survival in Merkel cell carcinoma. Cancer Lett. 2014, 344, 272–281. [Google Scholar] [CrossRef]
- Arora, R.; Shuda, M.; Guastafierro, A.; Feng, H.; Toptan, T.; Tolstov, Y.; Normolle, D.; Vollmer, L.L.; Vogt, A.; Dömling, A.; et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci. Transl. Med. 2012, 4, 133ra56. [Google Scholar] [CrossRef]
- Botić, T.; Defant, A.; Zanini, P.; Žužek, M.C.; Frangež, R.; Janussen, D.; Kersken, D.; Knez, Ž.; Mancini, I.; Sepčić, K. Discorhabdin alkaloids from Antarctic Latrunculia spp. sponges as a new class of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 136, 294–304. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; McCarthy, P.J.; Longley, R.E.; Pomponi, S.A.; Wright, A.E.; Lobkovsky, E.; Clardy, J. Discorhabdin P, a new enzyme inhibitor from a deep-water Caribbean sponge of the genus Batzella. J. Nat. Prod. 1999, 62, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Amusengeri, A.; Tastan Bishop, Ö. Discorhabdin N, a South African Natural Compound, for Hsp72 and Hsc70 Allosteric Modulation: Combined Study of Molecular Modeling and Dynamic Residue Network Analysis. Molecules 2019, 24, 188. [Google Scholar] [CrossRef] [PubMed]
- Goey, A.K.; Chau, C.H.; Sissung, T.M.; Cook, K.M.; Venzon, D.J.; Castro, A.; Ransom, T.R.; Henrich, C.J.; McKee, T.C.; McMahon, J.B.; et al. Screening and Biological Effects of Marine Pyrroloiminoquinone Alkaloids: Potential Inhibitors of the HIF-1α/p300 Interaction. J. Nat. Prod. 2016, 79, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.M.; Strope, J.D.; Beedie, S.L.; Huang, P.A.; Goey, A.K.L.; Cook, K.M.; Schofield, C.J.; Chau, C.H.; Cadelis, M.M.; Copp, B.R.; et al. Preclinical Evaluation of Discorhabdins in Antiangiogenic and Antitumor Models. Mar. Drugs 2018, 16, 241. [Google Scholar] [CrossRef]
- Grkovic, T.; Kaur, B.; Webb, V.L.; Copp, B.R. Semi-synthetic preparation of the rare, cytotoxic, deep-sea sourced sponge metabolites discorhabdins P and U. Bioorgan. Med. Chem. Lett. 2006, 16, 1944–1946. [Google Scholar] [CrossRef]
- Nyquist, R.A. (Ed.) Chapter 5—Sulfoxides, Sulfones, Sulfates, Monothiosulfates, Sulfonyl Halides, Sulfites, Sulfonamides, Sulfonates, and N-Sulfinyl Anilines. In Interpreting Infrared, Raman, and Nuclear Magnetic Resonance Spectra; Academic Press: San Diego, CA, USA, 2001; pp. 85–117. [Google Scholar]
- Schreiber, K.C. Infrared Spectra of Sulfones and Related Compounds. Anal. Chem. 1949, 21, 1168–1172. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ushio, M.; Kubota, T.; Yamamoto, S.; Fromont, J.; Kobayashi, J.I. Nakijiquinones J−R, Sesquiterpenoid Quinones with an Amine Residue from Okinawan Marine Sponges. J. Nat. Prod. 2010, 73, 467–471. [Google Scholar] [CrossRef]
- Avilov, S.A.; Kalinovsky, A.I.; Kalinin, V.I.; Stonik, V.A.; Riguera, R.; Jiménez, C. Koreoside A, a New Nonholostane Triterpene Glycoside from the Sea Cucumber Cucumaria koraiensis. J. Nat. Prod. 1997, 60, 808–810. [Google Scholar] [CrossRef]
- Guo, Q.; Xia, H.; Wu, Y.; Shao, S.; Xu, C.; Zhang, T.; Shi, J. Structure, property, biogenesis, and activity of diterpenoid alkaloids containing a sulfonic acid group from Aconitum carmichaelii. Acta Pharm. Sin. B 2020, 10, 1954–1965. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; McCombs, J.D.; Munro, M.H.G. Discorhabdin C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J. Org. Chem. 1986, 51, 5476–5478. [Google Scholar] [CrossRef]
- Yang, A.; Baker, B.J.; Grimwade, J.; Leonard, A.; McClintock, J.B. Discorhabdin Alkaloids from the Antarctic Sponge Latrunculia apicalis. J. Nat. Prod. 1995, 58, 1596–1599. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Zuleta, I.A.; Longley, R.E.; Wright, A.E.; Pomponi, S.A. Discorhabdins S, T, and U, New Cytotoxic Pyrroloiminoquinones from a Deep-Water Caribbean Sponge of the Genus Batzella. J. Nat. Prod. 2003, 66, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Antunes, E.M.; Beukes, D.R.; Kelly, M.; Samaai, T.; Barrows, L.R.; Marshall, K.M.; Sincich, C.; Davies-Coleman, M.T. Cytotoxic Pyrroloiminoquinones from Four New Species of South African Latrunculid Sponges. J. Nat. Prod. 2004, 67, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shi, X.; Yan, Q.; Li, Z.; Wang, Z.; Yu, H.; Wang, X.; Qi, J.; Jiang, M. Free-radical initiated cascade methylation or trideuteromethylation of isocyanides with dimethyl sulfoxides. RSC Adv. 2017, 7, 38830–38833. [Google Scholar] [CrossRef]
- Fallahi-Sichani, M.; Honarnejad, S.; Heiser, L.M.; Gray, J.W.; Sorger, P.K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9, 708–714. [Google Scholar] [CrossRef]
- Lam, C.F.; Grkovic, T.; Pearce, A.N.; Copp, B.R. Investigation of the electrophilic reactivity of the cytotoxic marine alkaloid discorhabdin B. Org. Biomol. Chem. 2012, 10, 3092–3097. [Google Scholar] [CrossRef]
- Lam, C.F.; Cadelis, M.M.; Copp, B.R. Exploration of the influence of spiro-dienone moiety on biological activity of the cytotoxic marine alkaloid discorhabdin P. Tetrahedron 2017, 73, 4779–4785. [Google Scholar] [CrossRef]
- Wada, Y.; Harayama, Y.; Kamimura, D.; Yoshida, M.; Shibata, T.; Fujiwara, K.; Morimoto, K.; Fujioka, H.; Kita, Y. The synthetic and biological studies of discorhabdins and related compounds. Org. Biomol. Chem. 2011, 9, 4959–4976. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Molnár, T.; Mázló, A.; Tslaf, V.; Szöllősi, A.G.; Emri, G.; Koncz, G. Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis. 2019, 10, 860. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2021, 163, 105297. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.K.; Chang, W.T.; Lin, I.L.; Chen, Y.F.; Padalwar, N.B.; Cheng, K.C.; Teng, Y.N.; Wang, C.H.; Chiu, C.C. The Role of Necroptosis in ROS-Mediated Cancer Therapies and Its Promising Applications. Cancers 2020, 12, 2185. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy inhibitors. Cell Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. From Merkel Cell Polyomavirus Infection to Merkel Cell Carcinoma Oncogenesis. Front. Microbiol. 2021, 12, 739695. [Google Scholar] [CrossRef] [PubMed]
- Grkovic, T.; Ding, Y.; Li, X.-C.; Webb, V.L.; Ferreira, D.; Copp, B.R. Enantiomeric Discorhabdin Alkaloids and Establishment of Their Absolute Configurations Using Theoretical Calculations of Electronic Circular Dichroism Spectra. J. Org. Chem. 2008, 73, 9133–9136. [Google Scholar] [CrossRef]
- Dijoux, M.-G.; Gamble, W.R.; Hallock, Y.F.; Cardellina, J.H.; van Soest, R.; Boyd, M.R. A New Discorhabdin from Two Sponge Genera. J. Nat. Prod. 1999, 62, 636–637. [Google Scholar] [CrossRef]
- Copp, B.R.; Fulton, K.F.; Perry, N.B.; Blunt, J.W.; Munro, M.H.G. Natural and Synthetic Derivatives of Discorhabdin C, a Cytotoxic Pigment from the New Zealand Sponge Latrunculia cf. bocagei. J. Org. Chem. 1994, 59, 8233–8238. [Google Scholar] [CrossRef]
- Grkovic, T.; Copp, B.R. New natural products in the discorhabdin A- and B-series from New Zealand-sourced Latrunculia spp. sponges. Tetrahedron 2009, 65, 6335–6340. [Google Scholar] [CrossRef]
- Jeon, J.-e.; Na, Z.; Jung, M.; Lee, H.-S.; Sim, C.J.; Nahm, K.; Oh, K.-B.; Shin, J. Discorhabdins from the Korean Marine Sponge Sceptrella sp. J. Nat. Prod. 2010, 73, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.H.; Dash, P.; Holland, P.; Kearsley, J.H.; Bell, J.R. Characterisation of four Merkel cell carcinoma adherent cell lines. Int. J. Cancer 1995, 60, 100–107. [Google Scholar] [CrossRef]
- Ronan, S.G.; Green, A.D.; Shilkaitis, A.; Huang, T.S.; Das Gupta, T.K. Merkel cell carcinoma: In vitro and in vivo characteristics of a new cell line. J. Am. Acad. Dermatol. 1993, 29 Pt 1, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.T.; Gould, V.E.; Salwen, H.R.; Herst, C.V.; Le Beau, M.M.; Lee, I.; Bauer, K.; Marder, R.J.; Andersen, R.; Kies, M.S.; et al. Establishment and characterization of a neuroendocrine skin carcinoma cell line. Lab. Investig. 1987, 56, 302–312. [Google Scholar] [PubMed]
- Martin, E.M.; Gould, V.E.; Hoog, A.; Rosen, S.T.; Radosevich, J.A.; Deftos, L.J. Parathyroid hormone-related protein, chromogranin A, and calcitonin gene products in the neuroendocrine skin carcinoma cell lines MKL1 and MKL2. Bone Miner. 1991, 14, 113–120. [Google Scholar] [CrossRef]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Duellman, S.J.; Zhou, W.; Meisenheimer, P.; Vidugiris, G.; Cali, J.J.; Gautam, P.; Wennerberg, K.; Vidugiriene, J. Bioluminescent, Nonlytic, Real-Time Cell Viability Assay and Use in Inhibitor Screening. Assay Drug Dev. Technol. 2015, 13, 456–465. [Google Scholar] [CrossRef]
- Niles, A.L.; Riss, T.L. Multiplexed viability, cytotoxicity, and caspase activity assays. Methods Mol. Biol. 2015, 1219, 21–33. [Google Scholar] [PubMed]
- Kupcho, K.; Shultz, J.; Hurst, R.; Hartnett, J.; Zhou, W.; Machleidt, T.; Grailer, J.; Worzella, T.; Riss, T.; Lazar, D.; et al. A real-time, bioluminescent annexin V assay for the assessment of apoptosis. Apoptosis 2019, 24, 184–197. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound 6 | Compound 9 | Compound 20 | |||
|---|---|---|---|---|---|---|
| δH Mult. (J Hz) | δC Mult. | δH Mult. (J Hz) | δC Mult. | δH Mult. (J Hz) | δC Mult. | |
| 1 | - | 160.1, C | 7.77 s | 150.1, CH | 7.86 s | 150.0, CH |
| 2 | 5.75 s | 125.3, CH | - | 122.0, C | - | 121.1, C |
| 3 | - | 179.3, C | - | 178.6, C | - | 170.9, C |
| 4 | 6.14 s | 118.5, CH | 6.61 s | 124.2, CH | - | 121.1, C |
| 5 | - | 167.8, C | - | 163.9, C | 7.86 s | 150.0, CH |
| 6 | - | 50.7, C | - | 46.3, C | - | 47.1 *, C |
| 7 | 2.59 dd (3.6,12.0) 2.93 m | 42.1, CH2 | 4.51 d (7.4) | 113.5, CH | 4.71 d (7.4) | 109.8, CH |
| 8 | 5.69 d (3.6) | 59.2, CH | 6.33 dd (5.0,7.4) | 124.6, CH | 6.53 dd (5.0,7.4) | 125.6, CH |
| 9 | 10.85 s | - | 10.38 d (5.0) | - | 10.71 d (5.0) | - |
| 10 | - | 151.5, C | - | 143.8, C | - | 145.7 *, C |
| 11 | - | 164.9, C | - | 166.7, C | - | 167.0, C |
| 12 | - | 123.7, C | - | 123.6, C | - | 123.7, C |
| 13 | 13.36 br d (2.0) | - | 13.22 br d (2.2) | - | 13.33 br d (2.0) | - |
| 14 | 7.42 d (2.0) | 127.6, CH | 7.37 d (2.8) | 126.9, CH | 7.41 d (2.8) | 127.2, CH |
| 15 | - | 120.5, C | - | 119.3, C | - | 119.6, C |
| 16 | 2.86 m | 17.7, CH2 | 2.82, m | 18.0, CH2 | 2.87, t (8.0) | 17.8, CH2 |
| 17 | 3.87 m 3.95 m | 44.9, CH2 | 3.61 m 3.75 m | 44.4, CH2 | 3.78 t (8.0) | 44.5, CH2 |
| 19 | - | 153.6, C | - | 158.0, C | - | 157.7, C |
| 20 | - | 97.6, C | - | 97.2, C | - | 95.4 *, C |
| 21 | - | 122.8, C | - | 122.7, C | - | 122.2, C |
| 1′ | - | 123.1, C | - | - | - | - |
| 3′ | 8.01 s | 141.2, CH | - | - | - | - |
| 5′ | - | 132.6, C | - | - | - | - |
| 6′ | 3.19 dd (9.6,15.2) 3.26 dd (6.0,15.2) | 24.2, CH2 | - | - | - | - |
| 7′ | 4.20 m | 50.9, CH | - | - | - | - |
| 8′ | - | 168.7, C | - | - | - | - |
| 9′ | 3.69 s | 32.3, CH3 | - | - | - | - |
| 10′ | 3.61 s | 53.0, CH3 | - | - | - | - |
| 11′ | 8.74 br s | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orfanoudaki, M.; Smith, E.A.; Hill, N.T.; Garman, K.A.; Brownell, I.; Copp, B.R.; Grkovic, T.; Henrich, C.J. An Investigation of Structure–Activity Relationships and Cell Death Mechanisms of the Marine Alkaloids Discorhabdins in Merkel Cell Carcinoma Cells. Mar. Drugs 2023, 21, 474. https://doi.org/10.3390/md21090474
Orfanoudaki M, Smith EA, Hill NT, Garman KA, Brownell I, Copp BR, Grkovic T, Henrich CJ. An Investigation of Structure–Activity Relationships and Cell Death Mechanisms of the Marine Alkaloids Discorhabdins in Merkel Cell Carcinoma Cells. Marine Drugs. 2023; 21(9):474. https://doi.org/10.3390/md21090474
Chicago/Turabian StyleOrfanoudaki, Maria, Emily A. Smith, Natasha T. Hill, Khalid A. Garman, Isaac Brownell, Brent R. Copp, Tanja Grkovic, and Curtis J. Henrich. 2023. "An Investigation of Structure–Activity Relationships and Cell Death Mechanisms of the Marine Alkaloids Discorhabdins in Merkel Cell Carcinoma Cells" Marine Drugs 21, no. 9: 474. https://doi.org/10.3390/md21090474
APA StyleOrfanoudaki, M., Smith, E. A., Hill, N. T., Garman, K. A., Brownell, I., Copp, B. R., Grkovic, T., & Henrich, C. J. (2023). An Investigation of Structure–Activity Relationships and Cell Death Mechanisms of the Marine Alkaloids Discorhabdins in Merkel Cell Carcinoma Cells. Marine Drugs, 21(9), 474. https://doi.org/10.3390/md21090474