The General Synthetic Methods were as follows: All air- or moisture-sensitive reactions were carried out in dried glassware (>100 °C) under an atmosphere of nitrogen or argon. Anhydrous solvents were purchased from Acros Organics (now Thermo Fisher Scientific, Waltham, MA, USA) or dried before use (THF was distilled over Na/benzophenone, diisopropylamine over CaH2, MeOH distilled with Magnesium and degassed via freeze–pump–thaw). ZnCl2 was fused in vacuo at 0.1 mbar prior to use. Ethyl acetate (EtOAc), petroleum ether and n-pentane were additionally distilled before use. Reactions that required cooling were cooled using conventional methods (ice/water for 0 °C, dry ice/acetone for −40 °C or −78 °C). Reactions that required heating above rt were heated using an oil bath. Reactions were monitored by NMR or analytical TLC, which was performed on precoated silica gel on Macherey-Nagel (Dueren, NRW, Germany) TLC-PET foils (Polygram® SIL G/UV254). Visualization was accomplished with UV-light (254 nm), KMnO4 solution or Ce(IV)/ammonium molybdate solution. The products were purified by flash chromatography on Macherey-Nagel 60 silica gel columns (0.063–0.2 mm or 0.04–0.063 mm) or by automated flash chromatography on Büchi (Flawil, Switzerland) Pure C-815 Chromatography System and prepacked Teledyne Isco (Thousand Oaks, CA, USA) silica gel cartridges (RediSep Rf normal- Macherey-Nagelphase silica flash 30–70 µm columns). Reversed-phase flash chromatography was accomplished by automated flash chromatography on Büchi Reveleris Prep Chromatography System and Büchi FlashPure Select C18 30 µm spherical cartridges or Cole-Parmer Telos (Vernon Hills, IL, USA) C18 cartridges. Preparative HPLC was performed on a Büchi Reveleris Prep Chromatography System using a Phenomenex (Danaher Corporation, Washington, DC, USA) Luna C18(2) 100 Å column (250 × 21.1 mm, 5 µm). 1H- and 13C-NMR spectra were recorded with a Bruker (Billerica, MA, USA) AV II 400 [400 MHz (1H), 100 MHz (13C)], a Bruker AV 500 [500 MHz, (1H), 125 MHz (13C)] or a Bruker Avance Neo 500 [500 MHz, (1H), 125 MHz (13C)] spectrometer in CDCl3 or DMSO-D6. NMR spectra were evaluated using NMR Processor Version 12.01 from Advanced Chemistry Development Inc. (ACD/Labs, Toronto, ON, Canada) or Bruker TopSpin Version 4.1.1. Chemical shifts are reported in ppm relative to Si(CH3)4 and the solvent residual peak was used as the internal standard. Selected signals for the minor diastereomers/rotamers are extracted from the spectra of the isomeric mixture. Multiplicities are reported as bs (broad signal), s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Signals marked with * in 13C-NMR give broad signals. Structural assignments were made with additional information from gCOSY, gNOESY, gHSQC, or gHMBC experiments. Melting points were determined with a melting-point apparatus MEL-TEMP II by Laboratory Devices (Auburn, CA, USA) and are uncorrected. High-resolution mass spectra (HRMS) were recorded with a Finnigan (now Thermo Fisher Scientific) MAT95 spectrometer using the CI technique (CI) or a Bruker Daltonics 4G hr-ToF (ESI-ToF) or a Bruker solariX using the ESI technique (ESI-FTICR). Other HRMS were measured on a Thermo Fisher Scientific Orbitrap Q exactive mass spectrometer, equipped with a heated ESI source and a Thermo Finnigan (Thermo Fisher Scientific) Ultimate3000 HPLC (ESI-Q exactive). Optical rotations were measured in CHCl3 with a Krüss (Hamburg, Germany) polarimeter P8000 T80 in thermostated (20 °C ± 1 °C) cuvettes and are given in 10−1degcm2g-1. The radiation source used was a sodium vapor lamp (λ = 589 nm). The concentrations are given in g/100 mL.
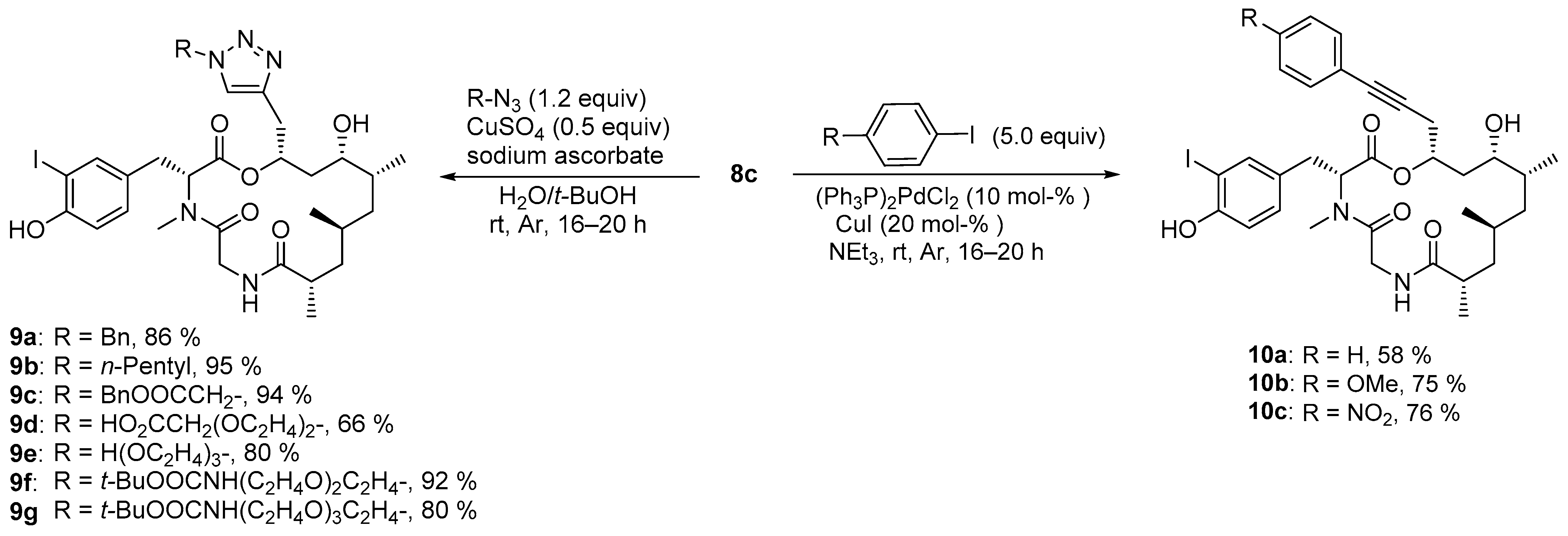
The general procedure for Cu(I)-catalyzed azide alkyne cycloadditions (CuAAC) was as follows: The alkyne (1.0 equiv) and azide (1.2 equiv) were dissolved in a 1:1 mixture of t-BuOH and H2O (0.05 M) in a 1.5 mL vial. Under a gentle stream of argon, sodium ascorbate (0.6 equiv, 1 M in H2O), and copper(II) sulfate (0.5 equiv, 1 M in H2O) were added, the vial sealed and the typically light-brownish suspension stirred overnight at rt (typically 16–20 h). The reaction mixture was concentrated and the residue purified by column chromatography.
The general procedure for Sonogashira cross-coupling of terminal alkyne was as follows: The alkyne (1.0 equiv), bis(triphenylphosphine)palladium(II) chloride (0.1 equiv), and copper(I) iodide (0.2 equiv) were added in a 1.5 mL vial and gently purged with argon for 5 min. Triethylamine (0.15 M) and the iodoarene (5.0 equiv) were added under a gentle stream of argon, the vial closed and the typically white suspension stirred overnight (typically 16–20 h) after which the reaction typically turned black. The suspension was diluted with MeCN, filtrated through a syringe filter (0.2 µm, PTFE), the solvent removed in vacuo, and the residue was subjected to chromatographic purification.
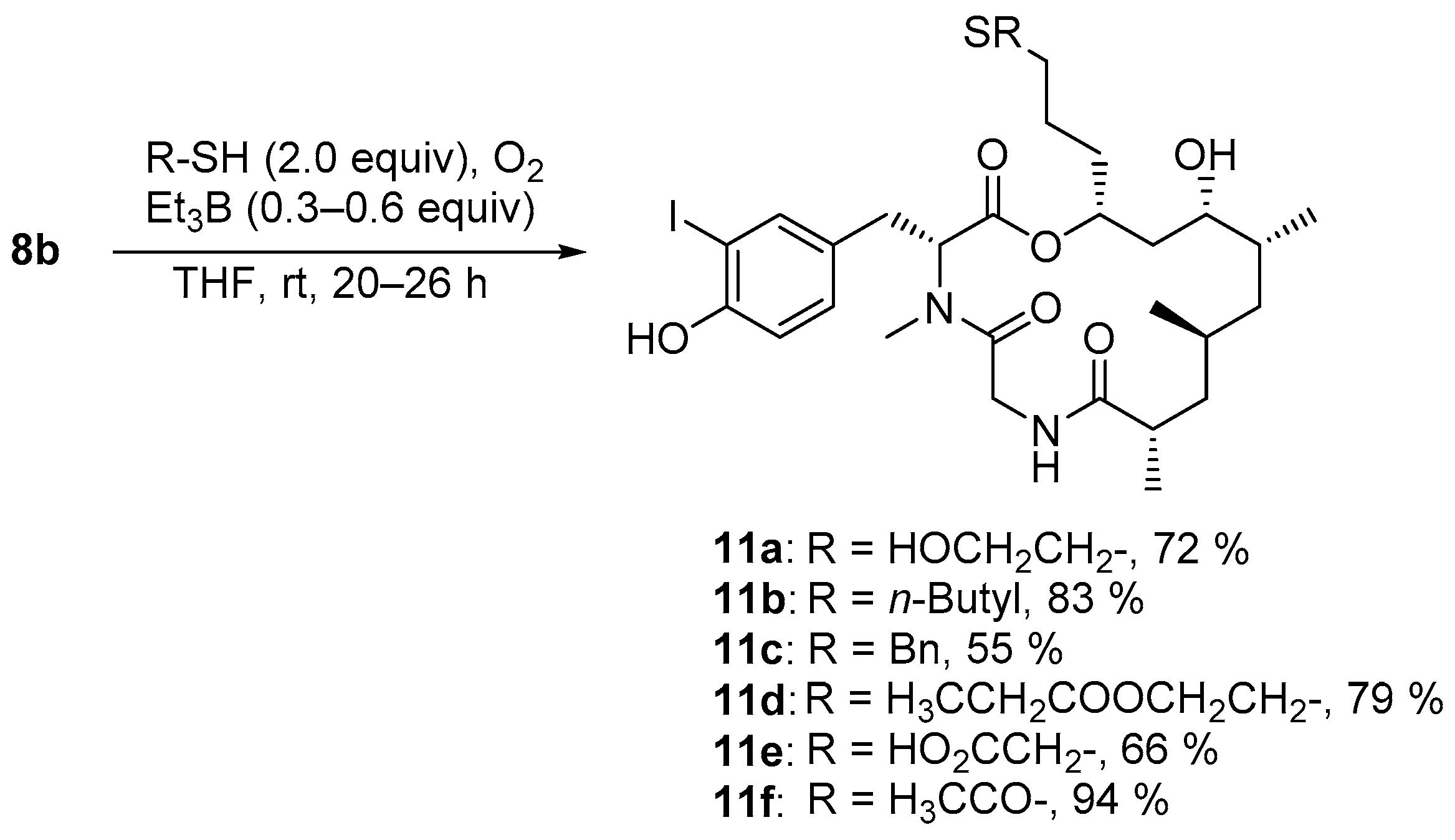
The general procedure for thiol-ene click reaction was as follows: The alkene (1.0 equiv) was gently purged with nitrogen and dissolved in THF abs. (0.1 M) in a 1.5 mL vial. Thiol (2.0 equiv) and Et3B (0.3 equiv, 1 M in hexane) were subsequently added, and the vial closed. Initiation of the reaction was performed by addition of air (0.4 mL) via a syringe and the reaction mixture stirred overnight at rt (typically 20–26 h). The solvent was removed and the crude product was purified by chromatography.
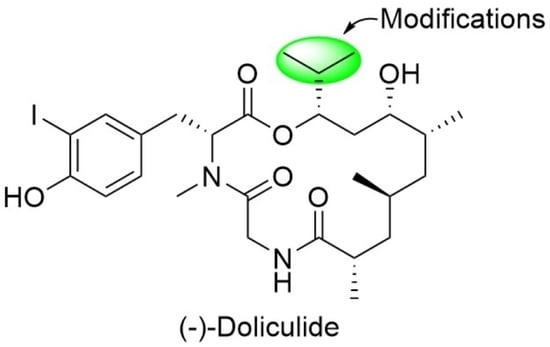
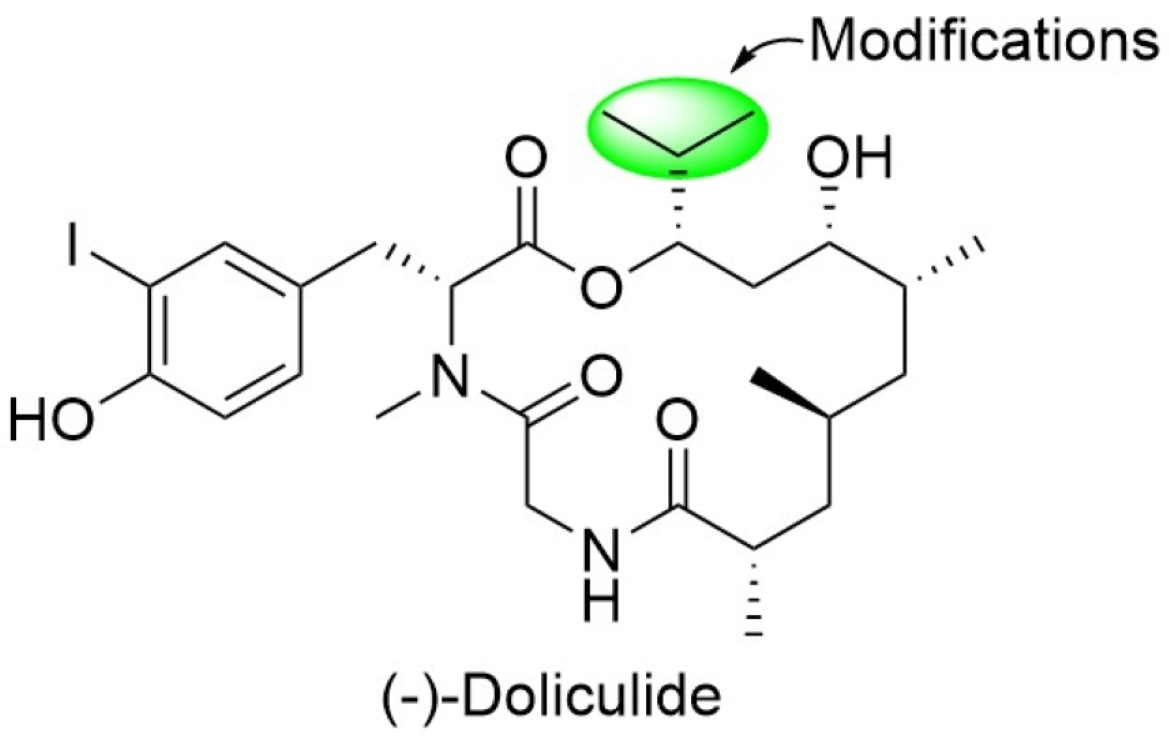
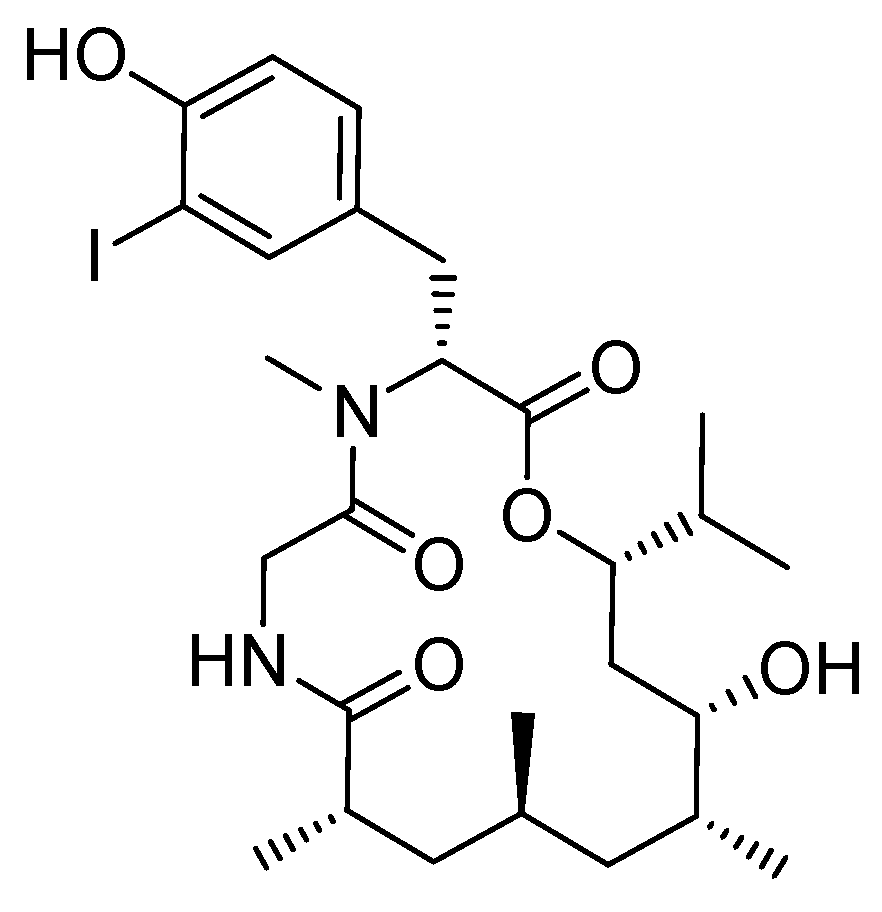
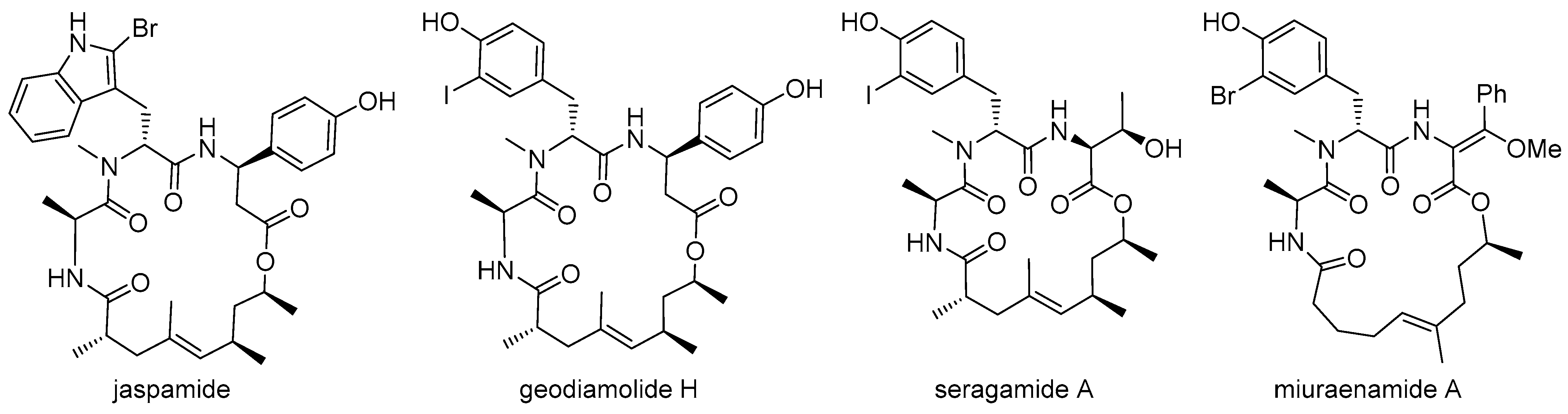
4.2. Syntheses of the Doliculide Core Structure
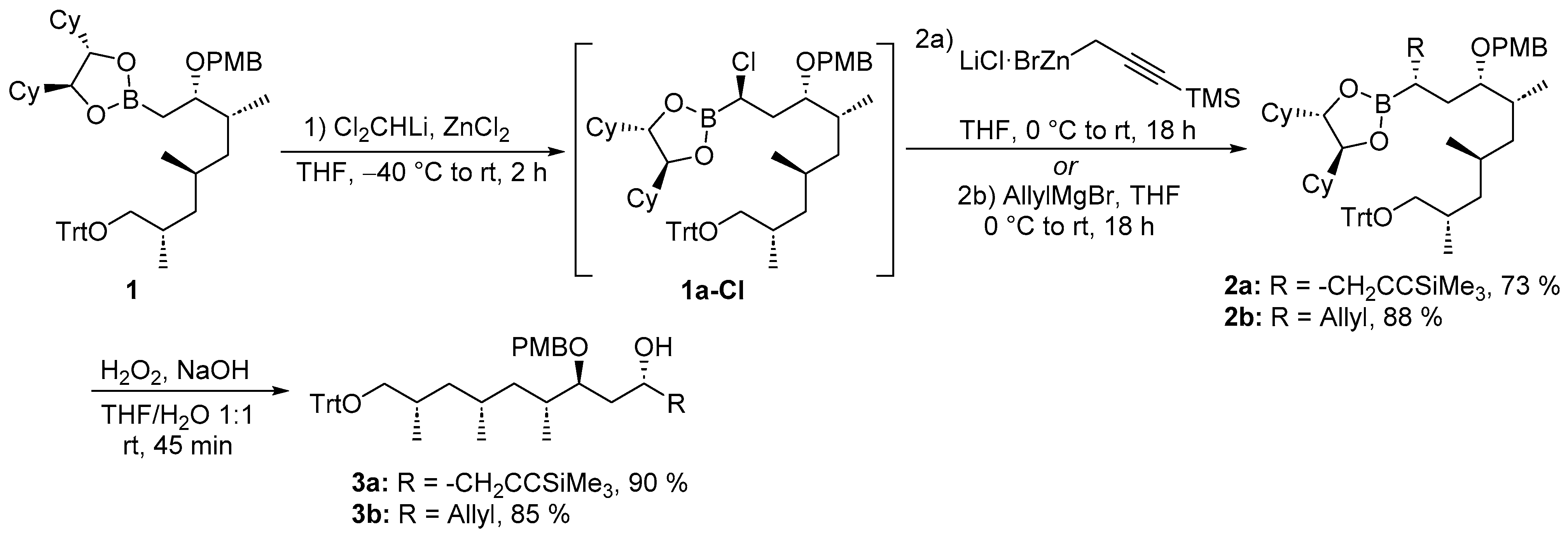
4.2.1. Synthesis of (4R,6S,7R,9R,11S)-6-[(4-Methoxybenzyl)oxy]-7,9,11-trimethyl-1-(trimethylsilyl)-12-(trityloxy)dodec-1-yn-4-yl (R)-3-[4-(allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoate (5a)
Carboxylic acid 4[30] (4.23 g, 9.18 mmol, 2.0 equiv) and alcohol 3a (3.27 g, 4.59 mmol, 1.0 equiv) were dissolved in anhydrous DCM (92.0 mL, 0.05 M) and cooled to −20 °C. DMAP (196 mg, 1.61 mmol, 0.35 equiv), EDC·HCl (1.76 g, 9.18 mmol, 2.0 equiv) and collidine (1.22 mL, 9.18 mmol, 2.0 equiv) were added and the reaction mixture stirred at -20 °C (acetone/cryostat) for 21 h. The reaction mixture was worked up with EtOAc and 1 M KHSO4. The organic phase was washed with saturated NaHCO3 and brine. The organic extract was dried over Na2SO4 and the solvent removed under reduced pressure. The residue was chromatographed (SiO2, petroleum ether:EtOAc 9:1–8:2) and ester 5a (4.88 g, 4.30 mmol, 94%) obtained as colorless resin. Rf (5a) = 0.44 (silica, petroleum ether: EtOAc 8:2). = −13.1 (c = 1.0, CHCl3). 1H-NMR (500 MHz, 373 K, DMSO-D6): δ = 0.13 (s, 9 H), 0.79 (d, J = 6.6 Hz, 3 H), 0.82 (d, J = 6.6 Hz, 3 H), 0.87–0.92 (m, 2 H), 1.18 (m, 1 H), 1.32 (s, 9 H), 1.37 (m, 1 H), 1.44 (m, 1 H), 1.70–1.78 (m, 3 H), 1.93 (m, 1 H), 2.56 (m, 2 H), 2.68 (s, 3 H), 2.85 (dd, J = 8.9, 6.6 Hz, 1 H), 2.91 (m, 1 H), 2.98 (dd, J = 8.9, 5.0 Hz, 1 H), 3.10 (dd, J = 14.4, 5.0 Hz, 1 H), 3.30 (m, 1 H), 3.75 (s, 3 H), 4.24 (d, J = 11.0 Hz, 1 H), 4.40 (d, J = 11.0 Hz, 1 H), 4.57 (m, 2 H), 4.69 (dd, J = 10.4, 5.0 Hz, 1 H), 5.08 (m, 1 H), 5.25 (dd, J = 1.6, 10.7 Hz, 1 H), 5.46 (dd, J = 1.6, 17.3 Hz, 1 H), 6.03 (ddt, J = 17.3, 10.7, 5.0 Hz, 1 H), 6.85–6.90 (m, 3 H), 7.14 (dd, J = 8.2, 2.2 Hz, 1 H), 7.17–7.26 (m, 5 H), 7.30 (m, 6 H), 7.39 (m, 6 H), 7.61 (d, J = 2.2 Hz, 1 H) ppm. 13C-NMR (125 MHz, 373 K, DMSO-D6): δ = −0.6, 13.8, 17.9, 20.5, 24.8, 27.5, 27.5, 30.7, 31.2, 31.2, 32.6, 32.7, 40.1, 40.8, 54.7, 59.6, 67.7, 69.1, 69.5, 69.8, 77.4, 78.7, 85.5, 86.0, 86.5, 102.4, 112.7, 113.3, 116.5, 126.3, 127.1, 127.8, 128.4, 129.5, 130.5, 131.7, 132.7, 138.7, 143.7, 154.0, 155.2, 158.4, 169.2 ppm. HRMS (ESI-ToF) calcd. for C44H67INO8Si+ [M–Trt+H]+: 892.3675 found 892.3674.
4.2.2. Synthesis of (4R,6S,7R,9R,11S)-6-[(4-Methoxybenzyl)oxy]-7,9,11-trimethyl-12-(trityloxy)dodec-1-en-4-yl (R)-3-[4-(allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoate (5b)
Carboxylic acid 4 (2.49 g, 5.39 mmol, 2.0 equiv) and alcohol 3b (1.67 g, 2.69 mmol, 1.0 equiv) were dissolved in anhydrous DCM (53.9 mL, 0.05 M) and cooled to -20 °C. DMAP (115 mg, 943 µmol, 0.35 equiv), EDC·HCl (1.03 g, 5.39 mmol, 2.0 equiv) and collidine (718 µL, 5.39 mmol, 2.0 equiv) were added and the reaction mixture stirred at -20 °C (acetone/cryostat) for 18 h. The reaction mixture was worked up with EtOAc and 1 M KHSO4. The organic phase was washed with saturated NaHCO3 and brine. The organic extract was dried over Na2SO4 and the solvent removed under reduced pressure. The residue was chromatographed (SiO2, petroleum ether:EtOAc 9:1–8:2) and ester 5b (2.41 g, 2.27 mmol, 84%) obtained as colorless resin. Rf (5b) = 0.35 (silica, n-pentane:EtOAc 8:2). = –8.0 (c = 1.0, CHCl3). 1H-NMR (500 MHz, 373 K, DMSO-D6): δ = 0.77 (d, J = 6.7 Hz, 3 H), 0.82 (d, J = 6.4 Hz, 3 H), 0.83–0.91 (m, 2 H), 0.93 (d, 3 H), 1.16 (m, 1 H), 1.30–1.36 (m, 10 H), 1.42 (m, 1 H), 1.58 (m, 2 H), 1.73 (m, 1 H), 1.91 (m, 1 H), 2.33 (m, 2 H), 2.65 (s, 3 H), 2.85 (dd, J = 9.0, 6.4 Hz, 1 H), 2.90 (dd, J = 14.3, 10.0 Hz, 1 H), 2.97 (dd, J = 9.0, 5.3 Hz, 1 H), 3.08 (dd, J = 14.3, 5.3 Hz, 1 H), 3.27 (ddd, J = 8.1, 8.1, 3.8 Hz, 1 H), 3.75 (s, 3 H), 4.24 (d, J = 11.1 Hz, 1H), 4.38 (d, J = 11.1 Hz, 1 H), 4.57 (ddd, J = 5.0, 1.6, 1.8 Hz, 2 H), 4.65 (dd, J = 10.0, 5.3 Hz, 1 H), 5.02–5.13 (m, 3 H), 5.25 (ddt, J = 10.4, 1.6, 1.6 Hz, 1 H), 5.45 (ddt, J = 17.2, 1.8, 1.6 Hz, 1 H), 5.73 (ddt, J = 17.1, 10.2, 7.0 Hz, 1 H), 6.02 (ddt, J = 17.3, 10.4, 5.1 Hz, 1 H), 6.84–6.90 (m, 3 H), 7.14 (dd, J = 8.4, 2.1 Hz, 1 H), 7.19 (d, J = 8.5 Hz, 2 H), 7.23 (tt, J = 7.3, 1.2 Hz, 3 H), 7.30 (m, 6 H), 7.40 (m, 6 H), 7.80 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, 373 K, DMSO-D6): δ = 13.8, 17.9, 20.5, 27.5, 27.5, 30.7, 31.1, 31.1, 32.6, 33.2, 38.1, 40.0, 40.8, 54.7, 59.6, 67.8, 69.1, 69.9, 71.2, 77.7, 78.7, 85.5, 86.0, 112.7, 113.3, 116.6, 117.0, 126.3, 127.1, 127.8, 128.4, 129.5, 130.5, 131.8, 132.7, 133.0, 138.6, 143.7, 154.0, 155.2, 158.4, 169.3 ppm. HRMS (ESI-Q exactive) calcd. for C60H74INO8Na+ [M+Na]+: 1086.4351 found 1086.4358.
4.2.3. Synthesis of (4R,6S,7R,9R,11S)-12-Hydroxy-6-[(4-methoxybenzyl)oxy]-7,9,11-trimethyl-1-(trimethylsilyl)dodec-1-yn-4-yl (R)-3-[4-(allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoate (5a-1)
Trityl ether 5a (2.52 g, 2.22 mmol, 1.0 equiv) was dissolved in MeOH (44.4 mL, 0.05 M). Amberlyst 15 (2.52 g, 100 m%) was added at rt and gently stirred for 20 h. EtOAc was added and the solid Amberlyst 15 was removed by filtration and stirred in ethyl acetate for 1 h. Amberlyst was again removed by filtration and both organic phases combined and washed with H2O. The aqueous phase was extracted with ethyl acetate and the combined organic extracts were dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was subjected to chromatographic purification (silica, n-pentane:EtOAc 7:3) and alcohol 5a-1 (1.69 g, 1.90 mmol, 85%) obtained as colorless resin. Rf (5a-1) = 0.33 (silica, n-pentane:EtOAc 7:3). = −16.6 (c = 0.5, CHCl3). 1H-NMR (500 MHz, 373 K, DMSO-D6): δ = 0.14 (s, 9 H), 0.83 (ddd, J = 13.8, 7.2, 7.2 Hz, 1 H), 0.85 (d, J = 6.6 Hz, 3 H), 0.86 (d, J = 6.9 Hz, 3 H), 0.89 (d, J = 6.6 Hz, 3 H), 0.94 (ddd, J = 13.8, 7.2, 7.2 Hz, 1 H), 1.23 (ddd, J = 13.8, 6.4, 6.4 Hz, 1 H), 1.30–1.37 (m, 10 H), 1.53–1.63 (m, 2 H), 1.71–1.77 (m, 2 H), 2.02 (m, 1 H), 2.54–2.60 (m, 2 H), 2.68 (s, 3 H), 2.91 (dd, J = 14.4, 10.4 Hz, 1 H), 3.11 (dd, J = 14.4, 5.0 Hz, 1 H), 3.16 (dd, J = 10.4, 6.6 Hz, 1 H), 3.30 (dd, J = 10.2, 5.2 Hz, 1 H), 3.33 (m, 1 H), 3.76 (s, 3 H), 4.27 (d, J = 11.3 Hz, 1 H), 4.44 (d, J = 11.3 Hz, 1 H), 4.58 (dt, J = 5.0, 1.6, 1.6 Hz, 2 H), 4.70 (dd, J = 10.4, 5.0 Hz, 1 H), 5.09 (m, 1 H), 5.26 (ddt, J = 10.6, 1.6, 1.6 Hz, 1 H), 5.46 (ddt, J = 17.3, 1.8, 1.8 Hz, 1 H), 6.03 (ddt, J = 17.3, 10.6, 4.9 Hz, 1 H), 6.87–6.92 (m, 3 H), 7.16 (dd, J = 8.5 Hz, J = 2.2 Hz, 1 H), 7.23 (m, 2 H), 7.61 (d, J = 2.2 Hz, 1 H) ppm. 13C-NMR (125 MHz, 373 K, DMSO-D6): δ = −0.5, 13.9, 17.4, 20.6, 24.9, 27.3, 27.5, 31.0, 31.0, 32.6, 32.6, 32.6, 40.2, 40.7, 54.6, 59.6, 66.0, 69.1, 59.6, 69.8, 77.2, 78.8, 86.0, 86.5, 102.5, 112.7, 113.3, 116.6, 128.5, 129.6, 130.5, 131.7, 132.8, 138.7, 155.2, 155.2, 158.4, 169.3 ppm. HRMS (ESI-ToF) calcd. for C44H67INO8Si+ [M+H]+: 892.3675 found 892.3660.
4.2.4. Synthesis of (4R,6S,7R,9R,11S)-12-Hydroxy-6-[(4-methoxybenzyl)oxy]-7,9,11-trimethyldodec-1-en-4-yl (R)-3-[4-(allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoate (5b-1)
Trityl ether 5b (2.32 g, 2.18 mmol, 1.0 equiv) was dissolved in MeOH (43.7 mL, 0.05 M). Amberlyst 15 (2.32 g, 100 m%) was added at rt and gently stirred for 20 h. EtOAc was added and the solid Amberlyst 15 was removed by filtration and stirred in ethyl acetate for 1 h. Amberlyst was again removed by filtration and both organic phases combined and washed with H2O. The aqueous phase was extracted with ethyl acetate and the combined organic extracts were dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was subjected to chromatographic purification (silica, n-pentane:EtOAc 7:3) and alcohol 5b-1 (1.61 g, 1.94 mmol, 89%) obtained as colorless resin. Rf (5b-1) = 0.33 (silica, n-pentane:EtOAc 7:3). = −11.6 (c = 1.0, CHCl3). 1H-NMR (500 MHz, 373 K, DMSO-D6): δ = 0.80–0.85 (m, J = 7.0 Hz, 4 H), 0.86 (d, J = 6.7 Hz, 3 H), 0.89 (d, J = 6.6 Hz, 3 H), 0.94 (m, 1 H), 1.23 (m, 1 H), 1.31–1-38 (m, 10 H), 1.52–1.64 (m, 4 H), 1.99 (m, 1 H), 2.35 (m, 2 H), 2.66 (s, 3 H), 2.92 (dd, J = 14.3, 10.3 Hz, 1 H), 3.10 (dd, J = 14.3, 5.3 Hz, 1 H), 3.17 (dd, J = 10.2, 6.6 Hz, 1 H), 3.30 (m, J = 10.2, 5.2 Hz, 2 H), 3.77 (s, 3 H), 4.27 (d, J = 11.1 Hz, 1 H), 2.42 (d, J = 11.1 Hz, 1 H), 4.58 (ddd, J = 4.9, 1.7, 1.5 Hz, 2 H), 4.67 (dd, J = 10.3, 5.3 Hz, 1 H), 5.02–5.13 (m, 3 H), 5.26 (ddt, J = 10.7, 1.5, 1.5 Hz, 1 H), 5.45 (ddt, J = 17.3, 1.7, 1.5 Hz, 1 H), 5.75 (ddt, J = 17.1, 10.2, 7.0 Hz, 1 H), 6.03 (ddt, J = 17.2, 10.5, 5.1 Hz, 1 H), 6.87–6.93 (m, 3 H), 7.17 (dd, J = 8.4, 2.0 Hz, 1 H), 7.23 (d, J = 8.5 Hz, 2 H), 7.62 (d, J = 8.5 Hz, 1 H) ppm. 13C-NMR (125 MHz, 373 K, DMSO-D6): δ = 13.9, 17.2, 20.5, 27.3, 27.5, 31.0, 32.0, 32.5, 32.6, 33.0, 38.1, 40.1, 40.7, 54.7, 59.7, 66.0, 69.1, 69.9, 71.2, 77.4, 78.8, 86.0, 112.7, 113.3, 116.6, 117.0, 128.5, 129.5, 130.6, 131.8, 132.8, 133.0, 138.7, 154.1, 155.2, 158.4, 169.3 ppm. HRMS (ESI-ToF) calcd. for C41H61INO8+ [M+H]+: 822.3436 found 822.3410.
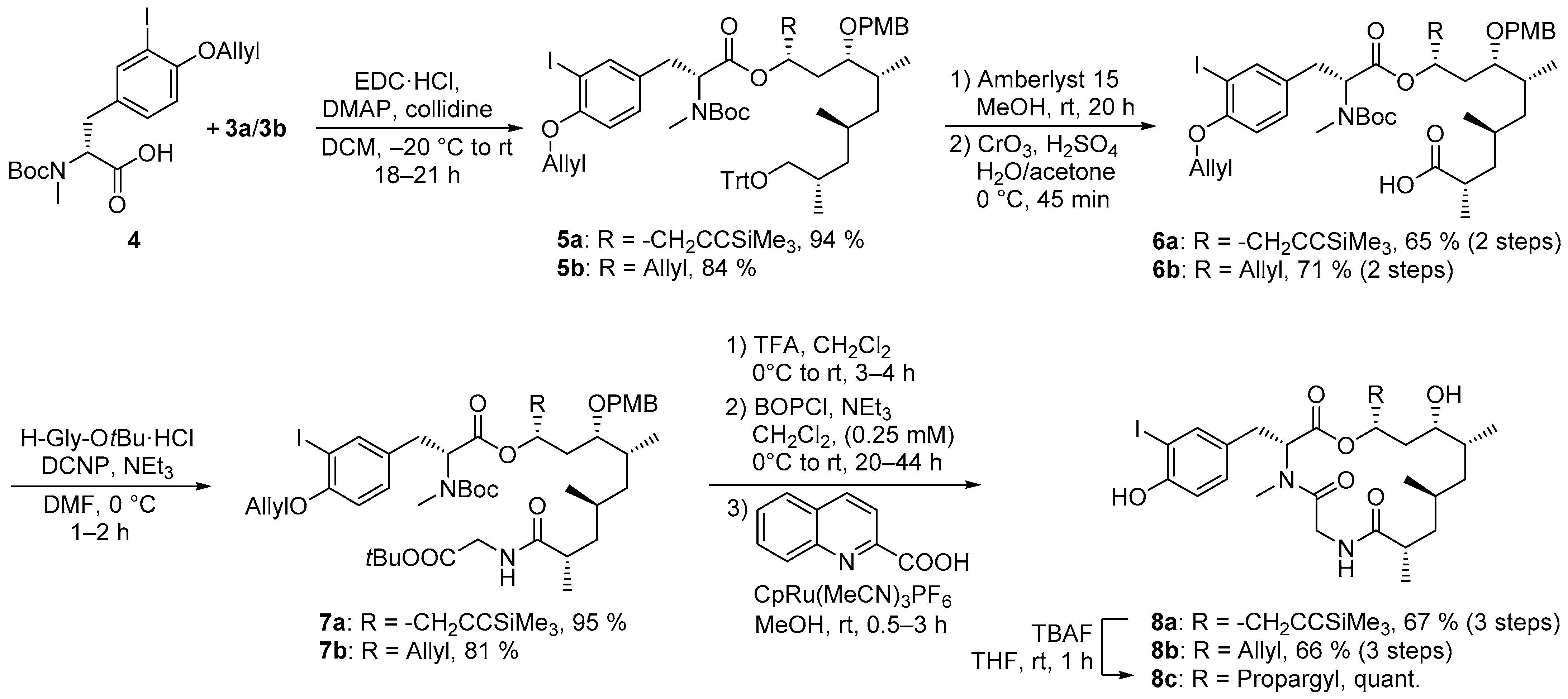
4.2.5. Synthesis of (2S,4S,6R,7S,9R)-9-({(R)-3-[4-(Allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoyl}oxy)-7-[(4-methoxybenzyl)oxy]-2,4,6-trimethyl-12-(trimethylsilyl)dodec-11-ynoic Acid (6a)
Alcohol 5a-1 (3.18 g, 3.57 mmol, 1.0 equiv) was dissolved in acetone (35.7 mL, 0.1 M) and cooled to 0 °C. Jones reagent (2.97 mL, 8.91 mmol, 3 M in 16% H2SO4, 2.5 equiv) were added and the mixture stirred for 45 min at this temperature. The reaction mixture was quenched with i-PrOH and concentrated under reduced pressure. The residue was diluted with H2O and EtOAc, the phases were separated and the aqueous phase extracted twice with EtOAc. The combined organic extracts were washed with brine, dried over Na2SO4. After removal of the solvent under reduced pressure, the residue chromatographically purified (silica, n-pentane:EtOAc 9:1–7:3) and acid 6a (2.47 g, 2.72 mmol, 76%) obtained as colorless resin. Rf (6a) = 0.33 (silica, n-pentane:EtOAc 7:3). = 0.6 (c = 0.5, CHCl3). 1H-NMR (500 MHz, 373 K, DMSO-D6): δ = 0.14 (s, 9 H), 0.85 (d, J = 6.7 Hz, 3 H), 0.90 (d, J = 6.6 Hz, 3 H), 0.95–1.05 (m, 2 H), 1.08 (d, J = 7.0 Hz, 3 H), 1.20 (ddd, J = 13.4, 8.0, 5.0 Hz, 1 H), 1.33 (s, 9 H), 1.54 (m, 1 H), 1.70 (ddd, J = 13.5, 9.0, 4.7 Hz, 1 H), 1.75 (m, 2 H), 1.98 (m, 1 H), 2.41 (ddq, J = 9.0, 7.1, 4.8 Hz, 1 H), 2.57 (m, 2 H), 2.69 (s, 3 H), 2.92 (dd, J = 14.4, 10.8 Hz, 1 H), 3.12 (dd, J = 14.4, 5.2 Hz, 1 H), 3.30 (ddd, J = 8.0, 4.0, 4.0 Hz, 1 H), 3.77 (s, 3 H), 4.29 (d, J = 11.1 Hz, 1 H), 4.44 (d, J = 11.1 Hz, 1 H), 4.59 (ddd, J = 5.0, 1.7, 1.5 Hz, 2 H), 4.71 (dd, J = 10.5, 5.2 Hz, 1 H), 5.08 (m, 1 H), 5.26 (ddt, J = 10.7, 1.5, 1.5 Hz, 1 H), 5.45 (ddt, J = 17.3, 1.7, 1.5 Hz, 1 H), 6.03 (ddt, J = 17.2, 10.5, 5.0 Hz, 1 H), 6.86–6.93 (m, 3 H), 7.16 (dd, J = 8.4, 2.0 Hz, 1 H), 7.23 (d, J = 8.5 Hz, 2 H), 7.62 (d, J = 2.0 Hz, 1 H). 13C-NMR (125 MHz, 373 K, DMSO-D6): δ = –0.46, 13.7, 17.6, 20.1, 25.0, 27.6, 28.1, 31.4, 31.4, 32.8, 33.0, 36.5, 40.2, 40.4, 54.9, 59.7, 69.3, 69.8, 70.0, 77.8, 79.0, 86.1, 86.7, 102.6, 112.9, 113.5, 116.8, 128.6, 129.7, 130.6, 131.9, 132.9, 138.8, 154.4, 155.4, 158.6, 169.5, 176.9. HRMS (ESI-ToF) calcd. for C44H65INO9Si+ [M+H]+: 906.3468 found 906.3487.
4.2.6. Synthesis of (2S,4S,6R,7S,9R)-9-({(R)-3-[4-(Allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoyl}oxy)-7-[(4-methoxybenzyl)oxy]-2,4,6-trimethyldodec-11-enoic Acid (6b)
Alcohol 5b-1 (1.51 g, 1.82 mmol, 1.0 equiv) was dissolved in acetone (18.2 mL, 0.1 M) and cooled to 0 °C. Jones reagent (1.51 mL, 4.54 mmol, 3 M in 16% H2SO4, 2.5 equiv) were added and the mixture stirred for 45 min at this temperature. The reaction mixture was quenched with i-PrOH and concentrated under reduced pressure. The residue was diluted with H2O and EtOAc, the phases were separated, and the aqueous phase was extracted twice with EtOAc. The combined organic extracts were washed with brine, dried over Na2SO4. After removal of the solvent under reduced pressure, the residue chromatographically purified (silica, n-pentane:EtOAc 9:1–7:3) and acid 6b (1.23 g, 1.46 mmol, 80%) obtained as colorless resin. Rf (6b) = 0.27 (silica, n-pentane:EtOAc 8:2). = –4.2 (c = 1.0, CHCl3). 1H-NMR (500 MHz, 373 K, DMSO-D6): δ = 0.83 (d, J = 6.9 Hz, 3 H), 0.90 (d, J = 6.6 Hz, 3 H), 0.97 (ddd, J = 13.5, 8.4, 6.1 Hz, 1 H), 1.02 (ddd, J = 13.7, 8.4, 5.3 Hz, 1 H), 1.08 (d, J = 6.9 Hz, 3 H), 1.19 (ddd, J = 13.5, 7.7, 5.3 Hz, 1 H), 1.35 (s, 9 H), 1.52 (m, 1 H), 1.61 (m, 2 H), 1.68 (ddd, J = 13.7, 8.9, 4.8 Hz, 1 H), 1.97 (m, 1 H), 2.28–2.38 ( (m, 2 H), 2.42 (ddq, J = 8.9, 6.9, 5.3 Hz, 1 H), 2.66 (s, 9 H), 2.92 (dd, J = 14.5, 10.1 Hz, 1 H), 3.11 (dd, J = 14.5, 5.5 Hz, 1 H), 3.28 (m, 1 H), 3.77 (s, 3 H), 4.27 (d, J = 11.1 Hz, 1 H), 4.42 (d, J = 11.1 Hz, 1 H), 4.59 (ddd, J = 4.9, 1.6, 1.6 Hz, 2 H), 4.68 (dd, J = 10.1, 5.4 Hz, 1 H), 5.03–5.13 (m, 3 H), 5.26 (ddt, J = 10.6, 1.6, 1.6 Hz, 1 H), 5.45 (ddt, J = 17.2, 1.6, 1.6 Hz, 1 H), 5.75 (ddt, J = 17.1, 10.2, 7.0 Hz, 1 H), 6.03 (ddt, J = 17.2, 10.5 Hz, J = 4.9 Hz, 1 H), 6.86–6.94 (m, 3 H), 7.17 (dd, J = 8.4, 2.0 Hz, 1 H), 7.23 (d, J = 8.5 Hz, 1 H), 7.63 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, 373 K, DMSO-D6): δ = 13.7, 17.4, 20.0, 27.5, 27.9, 31.2, 31.2, 32.6, 33.4, 36.4, 38.1, 39.9, 40.3, 54.7, 59.6, 69.1, 70.0, 71.2, 77.9, 78.9, 86.0, 112.7, 113.3, 116.6, 117.0, 128.5, 129.5, 130.6, 131.8, 132.8, 133.1, 138.7, 154.1, 155.2, 158.4, 169.3, 176.7 ppm. HRMS (ESI-ToF) calcd. for C41H59INO9+ [M+H]+: 836.3229 found 836.3228.
4.2.7. Synthesis of (4R,6S,7R,9S,11S)-12-{[2-(tert-Butoxy)-2-oxoethyl]amino}-6-[(4-methoxybenzyl)oxy]-7,9,11-trimethyl-12-oxo-1-(trimethylsilyl)dodec-1-yn-4-yl (R)-3-[4-(allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoate (7a)
Acid 6a (2.42 g, 2.67 mmol, 1.0 equiv) and tert-butyl glycinate hydrochloride (595 mg, 3.55 mmol, 1.33 equiv) were dissolved in anhydrous DMF (13.3 mL, 0.2 M). NEt3 (930 µL, 6.67 mmol, 0.726 gml−1, 2.5 equiv) and diethyl cyanophosphonate (DCNP) (1.03 mL, 6.14 mmol, 1.075 gml−1, 2.3 equiv) were added at 0 °C. After 1 h, brine and diethyl ether were added and the aqueous phase extracted twice with diethyl ether. The combined organic extracts were dried over Na2SO4 and the solvent removed under reduced pressure. After chromatographic purification (silica, n-pentane:EtOAc 8:2–7:3), amide 7a (2.57 g, 2.52 mmol, 95%) was obtained as colorless resin. Rf (7a) = 0.33 (silica, n-pentane:EtOAc 7:3). = −9.9 (c = 1.0, CHCl3). Major rotamer: 1H-NMR (500 MHz, CDCl3): δ = 0.14 (s, 9 H), 0.84 (m, 3 H), 0.90 (d, J = 6.3 Hz, 3 H), 0.99 (m, 2 H), 1.10 (m, 1 H), 1.38 (s, 9 H), 1.43–1.52 (m, 10 H), 1.65–1.85 (m, 3 H), 2.03 (m, 1 H), 2.38 (m, 1 H), 2.55 (m, 2 H), 2.68 (s, 3 H), 2.87 (dd, J = 14.2, 10.5 Hz, 1 H), 3.08–3.28 (m, 2 H), 3.79 (s, 3 H), 3.91 (m, 2 H), 4.23 (m, 1 H), 4.41–4.55 (m, 3 H), 4.73 (dd, J = 10.5, 5.4 Hz, 1 H), 5.19 (m, 1 H), 5.28 (m, 1 H), 5.47 (m, 1 H), 5.95–5.92 (m, 2 H), 6.78 (d, J = 8.5 Hz, 1 H), 6.87 (m, 2 H), 7.12 (d, J = 7.9 Hz, 1 H), 7.26 (m, 2 H), 7.60 (s, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 0.0, 14.0, 19.0, 21.1, 25.9, 28.0, 28.3, 41.7, 32.3, 33.5, 33.5, 33.9, 39.0, 41.0, 41.0, 41.9, 55.3, 59.9, 69.9, 70.7, 71.0, 78.6, 79.9, 82.0, 86.5, 87.2, 102.4, 112.5, 113.8, 117.5, 129.5, 129.9, 131.0, 132.0, 132.7, 139.8, 155.6, 156.0, 159.2, 169.2, 170.3, 176.4 (C-1) ppm. Minor rotamer: (selected signals) 1H-NMR (500 MHz, CDCl3): δ = 1.31 (s, 9 H), 2.74 (s, 3 H), 3.79 (s, 3 H), 7.03 (d, J = 7.9 Hz, 1 H), 7.61 (s, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 13.8, 28.5, 31.2, 60.6, 70.4, 80.2, 86.5, 87.6, 130.8, 139.8, 154.9, 156.1, 170.2, 176.3 ppm. HRMS (ESI-ToF) calcd. for C50H75IN2O10Si+ [M+H]+: 1019.4308 found 1019.4310.
4.2.8. Synthesis of (4R,6S,7R,9S,11S)-12-{[2-(tert-Butoxy)-2-oxoethyl]amino}-6-[(4-methoxybenzyl)oxy]-7,9,11-trimethyl-12-oxododec-1-en-4-yl (R)-3-[4-(allyloxy)-3-iodophenyl]-2-[(tert-butoxycarbonyl)(methyl)amino]propanoate (7b)
Acid 6b (1.19 g, 1.31 mmol, 1.0 equiv) and tert-butyl glycinate hydrochloride (293 mg, 1.75 mmol, 1.33 equiv) were dissolved in anhydrous DMF (6.56 mL, 0.2 M). NEt3 (457 µL, 3.28 mmol, 0.726 gml−1, 2.5 equiv) and diethyl cyanophosphonate (DCNP) (509 µL, 1.06 mmol, 1.075 gml−1, 2.3 equiv) were added at 0 °C. After 2 h, brine and diethyl ether were added and the aqueous phase extracted twice with diethyl ether. The combined organic extracts were dried over Na2SO4 and the solvent removed under reduced pressure. After chromatographic purification (silica, n-pentane:EtOAc 8:2), amide 7b (1.01 g, 1.06 mmol, 81%) was obtained as colorless resin. Rf (7b) = 0.43 (silica, n-pentane:EtOAc 7:3). = –4.2 (c = 1.0, CHCl3). 1H-NMR (500 MHz, 373 K, DMSO-D6): δ = 0.82 (d, J = 6.7 Hz, 3 H), 0.90 (d, J = 6.6 Hz, 3 H), 0.91–0.98 (m, 2 H), 1.04 (d, J = 6.9 Hz, 3 H), 1.16 (ddd, J = 13.4, 8.0, 5.0 Hz, 1 H), 1.35 (s, 9 H), 1.42 (s, 9 H), 1.48 (m, 1 H), 1.61 (m, 2 H), 1.71 (ddd, J = 13.5, 9.3, 4.3 Hz, 1 H), 1.97 (m, 1 H), 2.34 (m, 2 H), 2.43 (ddq, J = 9.3, 6.9, 5.0 Hz, 1 H), 2.67 (s, 3 H), 2.92 (dd, J = 14.3, 10.5 Hz, 1 H), 3.11 (dd, J = 14.3, 5.5 Hz, 1 H), 3.27 (m, 1 H), 3.67 (dd, J = 17.1, 6.0 Hz, 1 H), 3.72 (dd, J = 17.1, 6.0 Hz, 1 H), 3.77 (s, 3 H), 4.27 (d, J = 11.1 Hz, 1 H), 4.43 (d, = 11.1 Hz, 1 H), 4.58 (ddd, J = 4.9, 1.6, 1.6 Hz, 2 H), 4.69 (dd, J = 10.5, 5.5 Hz, 1 H), 5.02–5.13 (m, 3 H), 5.26 (ddt, J = 10.6, 1.6, 1.6 Hz, 1 H), 5.45 (ddt, J = 17.3, 1.8, 1.6 Hz, 1 H), 5.75 (ddt, J = 17.1, 10.2, 7.0 Hz, 1 H), 6.03 (ddt, J = 17.3, 10.5, 5.1 Hz, 1 H), 6.86–6.92 (m, 3 H), 7.17 (dd, J = 8.4, 2.1 Hz, 1 H), 7.23 (d, J = 8.7 Hz, 2 H), 7.63 (d, J = 2.0 Hz, 1 H), 7.71 (dd, J = 6.0, 6.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, 373 K, DMSO-D6): δ = 13.6, 18.1, 20.2, 27.3, 27.5, 27.6, 31.2, 31.2, 32.6, 33.4, 37.0, 38.1, 40.2, 40.4, 41.0, 54.7, 59.6, 69.1, 70.0, 71.2, 78.1, 78.7, 79.8, 85.9, 112.7, 113.3, 116.5, 117.0, 128.5, 129.5, 130.6, 131.8, 132.7, 133.1, 138.7, 154.1, 155.2, 158.4, 168.4, 169.3, 175.4 ppm. HRMS (ESI-ToF) calcd. for C47H70IN2O10+ [M+H]+: 949.4070 found 949.4059.
4.2.9. Synthesis of (3R,9S,11S,13R,14S,16R)-3-[4-(Allyloxy)-3-iodobenzyl]-14-hydroxy-4,9,11,13-tetramethyl-16-[3-(trimethylsilyl)prop-2-yn-1-yl]-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (7a-1)
A solution of linear precursor 7a (874 mg, 858 µmol, 1.0 equiv) in anhydrous DCM (3.86 mL, 4.5 mL/mmol, 0.22 M) was treated with TFA (2.57 mL, 3.0 mL/mmol, 0.33 M). It was warmed to rt and stirred for 90 min. The solvent was removed in N2 stream and azeotropically distilled with benzene and dried in vacuo. Triethylamine (1.20 mL, 8.58 mmol, 10.0 equiv) and BOP-Cl (1.09 g, 4.29 mmol, 5.0 equiv) were dissolved in DCM (1.72 l, 0.5 mM) and cooled to 0 °C. The previously obtained residue was dissolved in DCM (172 mL, 200 mL/mmol) and added dropwise (overnight) to the solution at 0 °C. After complete addition, the mixture was warmed to rt and further stirred for 24 h. The reaction mixture was worked up with 0.1 M HCl and the organic phase washed with saturated NaHCO3, saturated NH4Cl and brine. The organic phase was dried over Na2SO4 and the solvent removed under reduced pressure. The residue was dissolved in MeOH (51.5 mL, 60 mL/mmol, 0.017 M) and ammonia (4.29 mL, 5 mL/mmol, 35% in H2O) was added dropwise and stirred for 1 h. The solvent was removed under reduced pressure, the residue taken up on Isolute, reversed-phase chromatographed (Telos C18, H2O:MeCN 80:20–MeCN) and 7a-1 (480 mg, 662 µmol, 77%) obtained as colorless powder after lyophilization. A sample was further purified by preparative HPLC (Luna C18, H2O:MeCN 60:40–MeCN) for analytical purposes. Rf (7a-1) = 0.33 (silica, n-pentane:EtOAc 1:1). = −19.0 (c = 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ = 0.18 (s, 9 H), 0.83 (d, J = 6.7 Hz, 3 H), 0.95 (d, J = 6.1 Hz, 3 H), 0.98–1.09 (m, 3 H), 1.11 (d, J = 6.6 Hz, 3 H), 1.19 (m, 1 H), 1.37 (ddd, J = 14.3, 11.2, 1.7 Hz, 1 H), 1.45–1.58 (m, 2 H), 1.98 (m, 1 H), 2.24 (bs, 1 H), 2.38 (ddq, J = 12.0, 6.6, 3.3 Hz, 1 H), 2.54 (d, = 6.1 H, 2 H), 2.89 (dd, J = 15.5, 12.0 Hz, 1 H), 2.93 (s, 3 H), 3.27 (dd, J = 17.0, 1.0 Hz, 1 H), 3.42 (dd, J = 15.4, 4.5 Hz, 1 H), 3.61 (ddd, J = 11.2, 3.8, 1.7 Hz, 1 H), 4.55 (ddd, J = 4.8, 1.6, 1.6 Hz, 2 H), 4.77 (dd, J = 17.0, 8.6 Hz, 1 H), 5.26 (ddt, J = 11.4, 6.1, 1.8 Hz, 1 H), 5.30 (ddt, J = 10.6, 1.6, 1.3 Hz, 1 H), 5.47 (dd, J = 12.0, 4.5 Hz, 1 H), 5.50 (ddt, J = 17.1, 1.6, 1.3 Hz, 1 H), 6.03 (ddt, J = 17.2, 10.4, 5.0 Hz, 1 H), 6.21 (d, J = 8.1 Hz, 1 H), 6.71 (d, J = 8.4 Hz, 1 H), 7.09 (dd, J = 8.4, 2.0 Hz, 1 H), 7.59 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 0.0, 14.3, 17.6, 18.3, 26.4, 27.0, 30.5, 32.6, 33.1, 34.5, 39.0, 39.8, 42.7, 45.0, 57.8, 65.7, 69.7, 70.6, 86.8, 87.2, 102.0, 112.4, 117.7, 129.0, 130.7, 132.4, 139.1, 156.1, 171.0, 171.7, 177.4 ppm. HRMS (ESI-ToF) calcd. for C33H50IN2O6Si+ [M+H]+: 725.2477 found 725.2482.
4.2.10. Synthesis of (3R,9S,11S,13R,14S,16R)-16-allyl-3-[4-(allyloxy)-3-iodobenzyl]-14-hydroxy-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (7b-1)
A solution of linear precursor 7b (914 mg, 963 µmol, 1.0 equiv) in anhydrous DCM (4.33 mL, 4.5 mL/mmol, 0.22 M) was treated with TFA (2.89 mL, 3.0 mL/mmol, 0.33 M). It was warmed to rt and stirred for 90 min. The solvent was removed in N2 stream and azeotropically distilled with benzene and dried in vacuo. Triethylamine (1.34 mL, 9.63 mmol, 10.0 equiv) and BOP-Cl (1.23 g, 4.82 mmol, 5.0 equiv) were dissolved in DCM (1.93 l, 0.5 mM) and cooled to 0 °C. The previously obtained residue was dissolved in DCM (193 mL, 200 mL/mmol) and added dropwise (overnight) to the solution at 0 °C. After complete addition, the mixture was warmed to rt and further stirred for 24 h. The reaction mixture was worked up with 0.1 M HCl and the organic phase washed with saturated NaHCO3, saturated NH4Cl and brine. The organic phase was dried over Na2SO4 and the solvent removed under reduced pressure. The residue was dissolved in MeOH (57.8 mL, 60 mL/mmol, 0.017 M) and ammonia (4.82 mL, 5 mL/mmol, 35% in H2O) was added dropwise and stirred for 1 h. The solvent was removed under reduced pressure, the residue taken up on Isolute, reversed-phase chromatographed (FlashPure Select C18, H2O:MeCN 90:10–MeCN), and 7b-1 (511 mg, 781 µmol, 81%) obtained as colorless powder after lyophilization. A sample was further purified by preparative HPLC (Luna C18, H2O:MeCN 90:10–MeCN) for analytical purposes. = −33.7 (c = 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ = 0.82 (d, J = 6.7 Hz, 3 H), 0.95 (d, J = 6.1 Hz, 3 H), 0.98–1.08 (m, 3 H), 1.18 (m, 1 H), 1.31 (ddd, J = 13.9, 11.4, 2.0 Hz, 1 H), 1.42–1.55 (m, 2 H), 1.98 (m, 1 H), 2.07 (bs, 1 H), 2.35–2.43 (m, J = 6.7, 6.7 Hz, 3 H), 2.83 (dd, J = 15.3, 12.1 Hz, 1 H), 2.87 (s, 3 H), 3.24 (dd, J = 16.7, 1.0 Hz, 1 H), 3.38 (dd, J = 15.4, 4.5 Hz, 1 H), 3.60 (ddd, J = 11.4, 3.9, 1.8 Hz, 1 H), 4.54 (ddd, J = 4.9, 1.6, 1.6 Hz, 2 H), 4.78 (dd, J = 16.9, 8.7 Hz, 1 H), 5.04–5.14 (m, 2 H), 5.22–5.33 (m, J = 10.6, 1.5, 1.5 Hz, 2 H), 5.44 (dd, J = 12.2, 4.6 Hz, 1 H), 5.49 (dd, J = 17.3, 1.5, 1.5 Hz, 1 H), 5.74 (ddt, J = 17.0, 10.2, 6.9 Hz, 1 H), 6.03 (ddt, J = 17.2, 10.4, 5.0 Hz, 1 H), 6.24 (d, J = 8.3 Hz, 1 H), 6.70 (d, J = 8.4 Hz, 1 H), 7.08 (dd, J = 8.4, 2.0 Hz, 1 H), 7.58 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 14.3, 17.6, 18.3, 27.0, 30.6, 32.6, 33.2, 34.4, 39.0, 39.7, 39.8, 42.7, 44.9, 57.7, 65.6, 69.7, 71.7, 86.7, 112.4, 117.7, 118.0, 128.9, 130.7, 132.4, 133.8, 139.4, 156.1, 171.3, 171.6, 177.5 ppm. HRMS (ESI-ToF) calcd. for C30H44IN2O6+ [M+H]+: 655.2239 found 655.2247.
4.2.11. Synthesis of (3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-16-[3-(trimethylsilyl)prop-2-yn-1-yl]-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (8a)
0.01 M catalyst solution: CpRu(II)(MeCN)3PF6 (16.4 mg, 37.8 µmol) was dissolved in anhydrous MeOH (3.42 mL) and stirred for 5 min (yellow solution). Quinoline-2-carboxylic acid (380 µL, 38 µmol, 0.1 M in anhydrous MeOH) was added and the mixture stirred for 30 min (dark red solution). Deprotection: Allyl ether 7a-1 (413 mg, 569 µmol, 1.0 equiv) was dissolved in anhydrous degassed MeOH (11.4 mL, 0.05 M) under an N2-atmosphere. The previously prepared catalyst solution (2.85 mL, 28.5 µmol, 0.01 M) was added and the mixture stirred for 6 h at rt. The solvent was removed under reduced pressure, the residue taken up on Isolute and chromatographed (SiO2, n-pentane:EtOAc 1:1–4:6) and alcohol 8a (340 mg, 496 µmol, 87%) obtained as colorless powder after lyophilization. Rf (8a) = 0.13 (silica, n-pentane: EtOAc 1:1). = −20.1 (c = 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ = 0.18 (s, 9 H), 0.84 (d, J = 6.7 Hz, 3 H), 0.94 (d, J = 6.0 Hz, 3 H), 1.01–1.10 (m, 3 H), 1.12 (d, J = 6.6 Hz, 3 H), 1.15–1.23 (m, 1 H), 1.42 (m, 1 H), 1.48–1.60 (m, 2 H), 1.98 (m, 1 H), 2.31 (bs, 1 H), 2.43 (m, 1 H), 2.55 (m, 2 H), 2.87 (dd, J = 15.2, 12.0 Hz, 1 H), 2.97 (s, 3 H), 3.21 (d, J = 16.6 Hz, 1 H), 3.42 (dd, J = 15.2, 4.3 Hz, 1 H), 3.63 (m, 1 H), 4.78 (dd, J = 16.7, 8.7 Hz, 1 H), 5.27 (dddd, J = 10.2, 6.6, 6.6, 1.6 Hz, 1 H), 5.52 (dd, J = 11.9, 4.4 Hz, 1 H), 6.36 (d, J = 6.8 Hz, 1 H), 6.83 (d, J = 8.3 Hz, 1 H), 6.95 (bs, 1 H), 7.05 (dd, J = 8.3, 1.1 Hz, 1 H), 7.47 (d, J = 1.1 Hz, 1 H) ppm. 13C-NMR (100 MHz, CDCl3): δ = 0.0, 14.3, 17.6, 18.4, 26.4, 27.1, 30.8, 32.6, 33.0, 34.6, 39.1, 39.6, 42.8, 44.9, 57.8, 65.9, 70.7, 85.2, 87.3, 102.0, 115.2, 129.5, 130.2, 138.2, 154.3, 170.6, 172.1, 177.6 ppm. HRMS (ESI-ToF) calcd. for C30H46IN2O6Si+ [M+H]+: 685.2164 found 685.2162.
4.2.12. Synthesis of (3R,9S,11S,13R,14S,16R)-16-Allyl-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (8b)
0.01 M catalyst solution: CpRu(II)(MeCN)3PF6 (20.2 mg, 46.5 µmol) was dissolved in anhydrous MeOH (4.23 mL) and stirred for 5 min (yellow solution). Quinoline-2-carboxylic acid (470 µL, 470 µmol, 0.1 M in anhydrous MeOH) was added and the mixture stirred for 30 min (dark red solution). Deprotection: Allyl ether 7b-1 (463 mg, 707 µmol, 1.0 equiv) was dissolved in anhydrous degassed MeOH (14.1 mL, 0.05 M) under an N2-atmosphere. The previously prepared catalyst solution (3.53 mL, 35.3 µmol, 0.01 M) was added and the mixture stirred for 90 min at rt. The solvent was removed under reduced pressure, the residue taken up on Isolute and chromatographed (SiO2, n-pentane:EtOAc 1:1–2:8). The obtained residue was further purified by preparative HPLC (Luna C18, H2O:MeCN 50:50–MeCN) and compound 8b (354 mg, 576 µmol, 81%) obtained as colorless powder after lyophilization. Rf (8b) = 0.19 (silica, n-pentane:EtOAc 4:6). = −40.3 (c = 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3): δ = 0.83 (d, J = 6.9 Hz, 3 H), 0.96 (d, J = 6.1 Hz, 3 H), 1.00–1.10 (m, 3 H), 1.13 (d, J = 6.6 Hz, 3 H), 1.18 (m, 1 H), 1.25 (bs, 1 H), 1.34 (ddd, J = 14.0 Hz, J = 11.7, 2.0 Hz, 1 H), 1.48 (ddd, J= 14.0, 11.8, 2.0 Hz, 1 H), 1.52 (ddd, J = 12.1, 12.1, 1.5 Hz, 1 H), 1.99 (m, 1 H), 2.38 (dd, J = 7.0, 6.2 Hz, 2 H), 2.43 (ddd, J = 12.1, 6.6 Hz, 3.5 Hz, 1 H), 2.83 (dd, J = 15.4 Hz, J = 12.2 Hz, 1 H), 2.89 (s, 3 H), 3.23 (dd, J = 16.7, 1.6 Hz, 1 H), 3.40 (dd, J = 15.5, 4.5 Hz, 1 H), 3.62 (ddd, J = 11.6, 4.2, 2.1 Hz, 1 Hz), 4.80 (dd, J = 16.8, 8.7 Hz, 1 H), 5.06–5.15 (m, 2 H), 5.28 (ddt, J = 11.8, 6.2, 2.0 Hz, 1 H), 5.49 (dd, J = 12.1, 4.6 Hz, 1 H), 5.76 (ddt, J = 17.1, 10.2, 7.0 Hz, 1 H), 6.05 (bs, 1 H), 6.27 (d, J = 8.1 Hz, 1 H), 6.86 (d, J = 8.4 Hz, 1 H), 7.05 (dd, J = 8.3, 2.1 Hz, 1 H), 7.47 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.3, 17.6, 18.4, 27.0, 30.7, 32.7, 33.2, 34.5, 39.1, 39.6, 39.8, 42.9, 44.9, 57.8, 65.8, 71.8, 85.5, 115.2, 118.0, 129.7, 130.4, 133.8, 138.0, 154.0, 171.1, 171.8, 177.6 ppm. HRMS (ESI-ToF) calcd. for C27H40IN2O6+ [M+H]+: 615.1926 found 615.1944.
4.2.13. Synthesis of (3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-16-(prop-2-yn-1-yl)-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (8c)
Compound 8a (314 mg, 458 µmol, 1.0 equiv) was dissolved in anhydrous THF (9.17 mL, 0.05 M). TBAF (504 mL, 504 µmol, 1.1 equiv, 1 M solution in THF) was added at 0 °C and stirred for 15 min. The reaction was worked up with saturated NH4Cl and the aqueous phase extracted twice with EtOAc. The combined organic phases were dried over Na2SO4, and the solvent removed under reduced pressure. The residue was chromatographed (FlashPure Select C18, H2O:MeCN 90:10–MeCN) and alkyne 8c (281 mg, 458 µmol, quant.) obtained as colorless powder after lyophilization. Rf (8c) = 0.22 (silica, n-pentane: EtOAc 1:1). = −35.1 (c = 0.5, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.84 (d, J = 6.8 Hz, 3 H), 0.96 (d, J = 6.3 Hz, 3 H), 1.02–1.10 (m, 3 H), 1.12 (d, J = 6.7 Hz, 3 H), 1.20 (m, 1 H), 1.45–1.57 (m, 3 H), 1.98 (m, 1 H), 2.15 (t, J = 2.6 Hz, 1 H), 2.39–2.48 (m, 2 H), 2.61 (ddd, J = 17.1, 6.7, 2.6 Hz, 1 H), 2.88 (dd, J = 15.5, 11.9 Hz, 1 H), 2.94 (s, 3 H), 3.26 (d, J = 16.8 Hz, 1 H), 3.41 (dd, J = 15.5, 4.7 Hz, 1 H) 3.67 (m, 1 H), 4.81 (dd, J = 16.8, 8.7, 1 H), 5.26 (m, 1 H), 5.55 (dd, J = 11.9, 4.7 Hz, 1 H), 6.05 (bs, 1 H), 6.29 (m, 1 H), 6.86 (d, J = 8.2 Hz, 1 H), 7.06 (dd, J = 8.2, 2.0 Hz, 1 H), 7.49 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.3, 17.6, 18.3, 24.9, 27.0, 30.5, 32.4, 32.4, 34.7, 39.1, 39.8, 42.8, 45.0, 57.5, 65.9, 70.1, 71.4, 79.4, 85.5, 115.2, 129.8, 130.4, 138.1, 154.1, 170.6, 171.9, 177.5 ppm. HRMS (ESI-ToF) calcd. for C27H38IN2O6+ [M+H]+: 613.1769 found 613.1767.
4.3. Modification of the Doliculide Core Structure
4.3.1. Synthesis of (3R,9S,11S,13R,14S,16R)-16-[(1-Benzyl-1H-1,2,3-triazol-4-yl)methyl]-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (9a)
According to the general procedure for CuAAC, alkyne 8c (15.3 mg, 25.0 µmol, 1.0 equiv) was dissolved in a 1:1 mixture of t-BuOH:H2O (500 µL, 0.05 M) and reacted with benzyl azide (4.0 mg, 30.0 µmol, 1.2 equiv), sodium ascorbate (15.0 µL, 15.0 µmol, 1 M in H2O, 0.6 equiv), and copper(II) sulfate (12.5 µL, 12.5 µmol, 1 M in H2O, 0.5 equiv). After stirring for 16 h, the mixture was concentrated and the obtained crude product was subjected to reversed-phase flash chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN). Triazole 9a (17.5 mg, 23.5 µmol, 86%) was obtained as colorless powder after lyophilization. = –12.0 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.81 (d, J = 6.9 Hz, 3 H), 0.94 (d, J = 6.1 Hz, 3 H), 1.01–1.09 (m, 3 H), 1.13 (d, J = 6.7 Hz, 3 H), 1.17 (m, 1 H), 1.43–1.55 (m, 3 H), 1.97 (m, 1 H), 2.44 (m, 1 H), 2.61 (dd, J = 15.3, 12.1 Hz, 1 H), 2.78 (s, 3 H), 2.98–3.08 (m, 2 H), 3.09–3.17 (m, 2 H), 3.63 (ddd, J = 9.0, 4.1, 4.1 Hz, 1 H), 4.71 (dd, J = 16.7, 8.9 Hz), 5.37 (dd, J = 12.1, 4.6 Hz, 1 H), 5.44 (m, 1 H), 5.55 (d, J = 14.8 Hz, 1 H), 5.59 (d, J = 14.8 Hz, 1 H), 6.34 (dd, J = 8.9, 2.4 Hz, 1 H), 6.80 (d, J = 8.3 Hz, 1 H), 6.95 (dd, J = 8.2, 1.4 Hz, 1 H), 7.28–7.37 (m, 5 H), 7.39 (d, J = 1.7 Hz, 1 H), 7.43 (s, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.3, 17.6, 18.5, 27.0, 30.8, 31.5, 32.3, 33.1, 34.4, 39.1, 39.5, 42.9, 44.8, 54.0, 57.8, 65.8, 71.7, 85.1, 115.1, 121.9, 128.1, 128.7, 129.0, 129.4, 130.2, 135.0, 138.3, 143.9, 154.3, 171.1, 172.0, 177.7 ppm. HRMS (ESI-ToF) calcd. for C34H45IN5O6+ [M]+: 746.2409 found 746.2408.
4.3.2. Synthesis of (3R,9S,11S,13R,14S,16R)-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-16-((1-pentyl-1H-1,2,3-triazol-4-yl)methyl)-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (9b)
According to the general procedure for CuAAC, alkyne 8c (12.8 mg, 20.9 µmol, 1.0 equiv) was dissolved in a 1:1 mixture of t-BuOH:H2O (418 µL, 0.05 M) and reacted with 1-azidopentane (2.8 mg, 25.1 µmol, 1.2 equiv), sodium ascorbate (12.5 µL, 12.5 µmol, 1 M in H2O, 0.6 equiv) and copper(II) sulfate (10.5 µL, 10.5 µmol, 1 M in H2O, 0.5 equiv). After stirring for 16 h, the mixture was concentrated and the obtained crude product was subjected to reversed-phase flash chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN). Triazole 9b (14.4 mg, 19.8 µmol, 95%) was obtained as colorless powder after lyophilization. = −9.6 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.81 (d, J = 6.6 Hz, 3 H), 0.88 (t, J = 7.1 Hz, 3 H), 0.94 (d, J = 6.0 Hz, 3 H), 0.99–1.08 (m, 3 H), 1.11 (d, J = 6.6 Hz, 3 H), 1.19 (m, 1 H), 1.24–1.39 (m, 4 H), 1.42–1.56 (m, 3 H), 1.89 (tt, J = 7.3, 7.3 Hz, 2 H), 1.97 (m, 1 H), 2.43 (m, 1 H), 2.70 (dd, J = 14.3, 11.2 Hz, 1 H), 2.94 (s, 3 H), 2.98 (dd, J = 15.3, 3.8 Hz, 1 H), 3.06 (dd, J = 15.3, 8.2 Hz, 1 H), 3.11–3.21 (m, 2 H), 3.53–3.74 (m, 2 H), 4.36 (t, J = 7.3 Hz, 2 H), 4.76 (dd, J = 16.6, 8.7 Hz, 1 H), 5.33 (dd, J = 11.1, 3.9 Hz, 1 H), 5.44 (m, 1 H), 6.44 (bs, 1 H), 6.77 (d, J = 7.8 Hz, 1 H), 6.92 (d, J = 7.8 Hz, 1 H), 7.40 (s, 1 H), 7.43 (s, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 13.9, 14.4, 17.7, 18.4, 22.1, 27.0, 28.6, 30.1, 31.0, 31.6, 32.4, 33.2, 34.5, 39.0, 39.6, 42.8, 44.9, 50.3, 58.0, 65.8, 71.9, 86.0, 115.5, 121.7, 129.5, 129.5, 138.2, 143.5, 155.5, 171.3, 171.9, 177.6 ppm. HRMS (ESI-ToF) calcd. for C32H49IN5O6+ [M+H]+: 726.2722 found 726.2722.
4.3.3. Synthesis of Benzyl 2-(4-{[(3R,9S,11S,13R,14S,16R)-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-2,5,8-trioxo-1-oxa-4,7-diazacyclohexadecan-16-yl]methyl}-1H-1,2,3-triazol-1-yl)acetate (9c)
According to the general procedure for CuAAC, alkyne 8c (11.5 mg, 18.8 µmol, 1.0 equiv) was dissolved in a 1:1 mixture of t-BuOH:H2O (374 µL, 0.05 M) and reacted with benzyl 2-azidoacetate (4.3 mg, 22.5 µmol, 1.2 equiv), sodium ascorbate (11.3 µL, 11.3 µmol, 1 M in H2O, 0.6 equiv) and copper(II) sulfate (9.4 µL, 9.4 µmol, 1 M in H2O, 0.5 equiv). After stirring for 16 h, the mixture was concentrated and the obtained crude product was subjected to reversed-phase flash chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN). Triazole 9c (14.2 mg, 17.7 µmol, 94%) was obtained as colorless powder after lyophilization. = –13.0 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.82 (d, J = 6.7 Hz, 3 H), 0.94 (d, J = 6.0 Hz, 3 H), 0.99–1.07 (m, 3 H), 1.10 (d, J = 6.6 Hz, 3 H), 1.16 (m, 1 H), 1.44–1.51 (m, 2 H), 1.58 (m, 1 H), 2.0 (m, 1 H), 2.42 (ddq, J = 12.1, 6.6 Hz, J = 3.4 Hz, 1 H), 2.78 (dd, J = 15.4, 12.4 Hz, 1 H), 2.80 (s, 3 H), 3.05 (dd, J = 16.6, 2.0 Hz, 1 H), 3.09 (dd, J = 16.3, 6.4 Hz, 1 H), 3.23 (dd, J = 16.3, 3.8 Hz, 1 H), 3.26 (dd, J = 15.4, 4.4 Hz, 1 H), 3.68 (m, 1 H), 4.70 (dd, J = 16.6, 9.2 Hz, 1 H), 5.19 (d, J = 12.2 Hz, 1 H), 5.25 (d, J = 12.2 Hz, 1 H), 5.33 (d, J = 17.5 Hz, 1 H), 5.37 (d, J = 17.5 Hz, 1 H), 5.47 (dd, J = 12.2, 4.4 Hz, 1 H), 5.58 (m, 1 H), 6.13 (dd, J = 9.2, 2.0 Hz, 1 H), 6.81 (d, J = 8.2 Hz, 1 H), 7.01 (dd, J = 8.2, 1.4 Hz, 1 H), 7.29–7.39 (m, 5 H), 7.46 (d, J = 1.7 Hz, 1 H), 7.68 (s, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.2, 17.5, 18.5, 26.9, 30.7, 31.3, 32.2, 32.5, 34.2, 39.0 39.3, 43.1, 44.8, 50.8, 57.6, 65.8, 67.8, 71.1, 85.2, 115.1, 123.4, 128.4, 128.7, 128.7, 129.5, 130.4, 134.7, 138.3, 143.4, 154.2, 167.0, 171.2, 172.0, 177.9 ppm. HRMS (ESI-ToF) calcd. for C29H41IN5O8+ [M–Bn+2H]+: 714.1994 found 714.1991.
4.3.4. Synthesis of 2-{2-[2-(4-{[(3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-2,5,8-trioxo-1-oxa-4,7-diazacyclohexadecan-16-yl]methyl}-1H-1,2,3-triazol-1-yl)ethoxy]ethoxy}acetic Acid (9d)
According to the general procedure for CuAAC, alkyne 8c (9.9 mg, 16.2 µmol, 1.0 equiv) was dissolved in a 1:1 mixture of t-BuOH:H2O (324 µL, 0.05 M) and reacted with potassium 2-[2-(2-azidoethoxy)ethoxy)acetate (4.4 mg, 19.4 µmol, 1.2 equiv), sodium ascorbate (9.7 µL, 9.7 µmol, 1 M in H2O, 0.6 equiv) and copper(II) sulfate (8.1 µL, 8.1 µmol, 1 M in H2O, 0.5 equiv). After stirring for 18 h. DCM and brine were added to the reaction mixture, the phases separated and the aqueous phase extracted twice with DCM and once with CHCl3/i-PrOH (3:1). The combined organic extracts were concentrated and the obtained crude product was subjected to reversed-phase flash chromatography (FlashPure Select C18, H2O + 0.1% HCOOH:MeCN 90:10–MeCN). Triazole 9d (8.5 mg, 10.6 µmol, 66%) was obtained as colorless powder after lyophilization. = −4.1 (c = 1.0, CHCl3). 1H-NMR (500 MHz, DMSO-D6): δ = 0.68 (d, J = 6.9 Hz, 3 H), 0.84 (d, J = 6.3 Hz, 3 H), 0.87–0.92 (m, 3 H), 0.94 (d, J = 6.7 Hz, 3 H), 1.15 (m, 1 H), 1.22–1.38 (m, 3 H), 1.57 (bs, 1 H), 1.77 (m, 1 H), 2.47 (m, 1 H), 2.79 (dd, J = 15.0, 11.4 Hz, 1 H), 2.82 (s, 3 H), 2.92 (dd, J = 15.0, 4.6 Hz, 1 H), 2.98 (dd, J = 15.2, 6.0 Hz, 1 H), 3.01 (dd, J = 15.7, 3.1 Hz, 1 H), 3.09 (dd, J = 15.0, 4.6 Hz, 1 H), 3.34 (bs, 1 H), 3.51–3.58 (m, 5 H), 3.83 (t, J = 5.4 Hz, 2 H), 3.87 (s, 2 H), 4.49–4.60 (m, 3 H), 5.21 (dddd, J = 9.2, 6.0, 5.6, 4.6 Hz, 1 H), 5.38 (dd, J = 11.4, 4.6 Hz, 1 H), 6.78 (d, J = 8.2 Hz, 1 H), 6.97 (dd, J = 8.4, 2.0 Hz, 1 H), 7.47 (d, J = 2.0 Hz, 1 H), 7.93 (s, 1 H), 8.17 (dd, J = 8.6, 3.1 Hz, 1 H), 10.5 (bs, 1 H) ppm. 13C-NMR (125 MHz, DMSO-D6): δ = 14.4, 17.5, 18.6, 26.2, 30.5, 30.8, 31.0, 31.4, 34.5, 37.1, 38.6, 43.0, 44.7, 49.1, 56.6, 63.9, 68.3, 68.9, 69.5, 69.5, 70.8, 84.4, 114.7, 123.2, 129.6, 130.0, 138.4, 142.0, 155.2, 169.9, 170.7, 171.8, 176.2 ppm. HRMS (ESI-ToF) calcd. for C33H49IN5O10+ [M+H]+: 802.2519 found 802.2518.
4.3.5. Synthesis of (3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-16-((1-(2-(2-(2-hydroxyethoxy)ethoxy)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (9e)
According to the general procedure for CuAAC, alkyne 8c (14.8 mg, 24.2 µmol, 1.0 equiv) was dissolved in a 1:1 mixture of t-BuOH:H2O (484 µL, 0.05 M) and reacted with 2-[2-(2-azidoethoxy]ethoxy)ethan-1-ol (5.1 mg, 29.1 µmol, 1.2 equiv), sodium ascorbate (14.5 µL, 14.5 µmol, 1 M in H2O, 0.6 equiv) and copper(II) sulfate (12.1 µL, 12.1 µmol, 1 M in H2O, 0.5 equiv). After stirring for 16 h, the mixture was concentrated and the obtained crude product was subjected to reversed-phase flash chromatography (FlashPure Select C18, H2O + 0.1% HCOOH:MeCN 90:10–MeCN). Triazole 9e (15.2 mg, 19.3 µmol, 80%) was obtained as colorless powder after lyophilization. = –9.6 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.82 (d, J = 6.0 Hz, 3 H), 0.93 (d, J = 6.1 Hz, 3 H), 0.99–1.08 (m, 3 H), 1.12 (d, J = 6.6 Hz, 3 H), 1.17 (m, 1 H), 1.42–1.58 (m, 3 H), 1.88–2.07 (m, 3 H), 2.43 (ddq, J = 12.1, 6.6, 3.4 Hz, 1 H), 2.71 (dd, J = 15.3, 11.8 Hz, 1 H), 2.93 (s, 3 H), 2.99 (dd, J = 15.4, 4.1 Hz, 1 H), 3.07 (dd, J = 15.0, 7.9 Hz, 1 H), 3.13 (dd, J = 16.6, 1.5 Hz, 1 H), 3.17 (dd, J = 15.4, 4.6 Hz, 1 H), 3.58 (t, J = 4.4 Hz, 2 H), 3.60–3.65 (m, 5 H), 3.75 (dd, J = 4.1, 4.1 Hz, 2 H), 3.83–3.92 (m, 2 H), 4.57 (m, 2 H), 4.75 (dd, J = 16.6, 8.9 Hz, 1 H), 5.34 (dd, J = 11.8, 4.7 Hz, 1 H), 5.43 (m, 1 H), 6.41 (dd, J = 8.9, 1.5 Hz, 1 H), 6.82 (d, J = 8.1 Hz, 1 H), 6.97 (dd, J = 8.1, 1.7 Hz, 1 H), 7.44 (d, J = 1.7 Hz, 1 H), 7.73 (s, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.3, 17.6, 18.5, 27.0, 31.0, 31.5, 32.4, 33.3, 34.4, 39.1, 39.6, 42.9, 44.9, 50.1, 58.0, 61.6, 65.9, 69.5, 70.2, 70.4, 72.2, 72.5, 85.0, 115.2, 123.4, 129.6, 130.2, 138.4, 143.6, 154.4, 171.2, 171.9, 177.8 ppm. HRMS (ESI-ToF) calcd. for C33H51IN5O9+ [M+H]+: 788.2726 found 788.2727.
4.3.6. Synthesis of tert-Butyl (2-{2-[2-(4-{[(3R,9S,11S,13R,14S,16R)-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-2,5,8-trioxo-1-oxa-4,7-diazacyclohexadecan-16-yl]methyl}-1H-1,2,3-triazol-1-yl)ethoxy]ethoxy}ethyl)carbamate (9f)
According to the general procedure for CuAAC, alkyne 8c (10.2 mg, 16.7 µmol, 1.0 equiv) was dissolved in a 1:1 mixture of t-BuOH:H2O (334 µL, 0.05 M) and reacted with tert-butyl {2-[2-(2-aminoethoxy)ethoxy]ethyl}carbamate (5.5 mg, 20.0 µmol, 1.2 equiv), sodium ascorbate (10.0 µL, 10.0 µmol, 1 M in H2O, 0.6 equiv) and copper(II) sulfate (8.3 µL, 8.3 µmol, 1 M in H2O, 0.5 equiv). After stirring for 16 h, the mixture was concentrated and the obtained crude product was subjected to reversed-phase flash chromatography (FlashPure Select C18, H2O + 0.1% HCOOH:MeCN 90:10–MeCN). Triazole 9f (13.6 mg, 15.3 µmol, 92%) was obtained as colorless powder after lyophilization. = –4.3 (c = 1.0, CHCl3). 1H-NMR (500 MHz, DMSO-D6): δ = 0.68 (d, J = 6.7 Hz, 3 H), 0.84 (d, J = 6.1 Hz, 3 H), 0.86–0.93 (m, 3 H), 0.94 (d, J = 6.7 Hz, 3 H), 1.15 (m, 1 H), 1.22–1.35 (m, 3 H), 1.36 (s, 9 H), 1.77 (m, 1 H), 2.48 (m, 1 H), 2.80 (dd, J = 15.0, 11.7 Hz, 1 H), 2.81 (s, 3 H), 2.92 (dd, J = 15.1, 5.0 Hz, 1 H), 2.95–3.06 (m, 4 H), 3.09 (dd, J = 15.0, 4.3 Hz, 1 H), 3.34 (t, J = 6.0 Hz, 2 H), 3.45 (m, 2 H), 3.51 (m, 2 H), 3.57 (m, 1 H), 3.82 (ddd, J = 16.2, 10.8, 5.3 Hz, 1 H), 3.83 (ddd, J = 16.2, 11.1, 5.2 Hz, 1 H), 4.21 (d, J = 4.7 Hz, 1 H), 4.51–4.60 (m, 3 H), 5.21 (m, 1 H), 5.40 (dd, J = 11.5, 4.5 Hz, 1 H), 6.68–6.80 (m, 2 H), 6.98 (dd, J = 8.2, 1.8 Hz, 1 H), 7.47 (d, J = 1.8 Hz, 1 H), 7.91 (s, 1 H), 8.16 (dd, J = 8.6, 3.1 Hz, 1 H), 10.1 (bs, 1 H) ppm. 13C-NMR (125 MHz, DMSO-D6): δ = 14.4, 17.5, 18.6, 26.2, 28.2, 30.4, 30.8, 30.9, 31.3, 34.4, 37.1, 38.5, 39.7, 43.0, 44.7, 49.1, 56.5, 63.8, 68.0, 69.2, 69.4, 69.5, 70.7, 77.6, 84.3, 114.7, 123.2, 129.6, 130.1, 138.5, 141.9, 155.0, 155.6, 169.9, 170.7, 176.1 ppm. HRMS (ESI-ToF) calcd. for C38H60IN6O10+ [M+H]+: 887.3410 found 887.3397.
4.3.7. Synthesis of tert-Butyl [2-(2-{2-[2-(4-{[(3R,9S,11S,13R,14S,16R)-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-2,5,8-trioxo-1-oxa-4,7-diazacyclohexadecan-16-yl]methyl}-1H-1,2,3-triazol-1-yl)ethoxy]ethoxy}ethoxy)ethyl]carbamate (9g)
According to the general procedure for CuAAC, alkyne 8c (10.2 mg, 16.7 µmol, 1.0 equiv) was dissolved in a 1:1 mixture of t-BuOH:H2O (334 µL, 0.05 M) and reacted with tert-butyl (2-{2-[2-(2-aminoethoxy)ethoxy]ethoxy}ethyl)carbamate (6.4 mg, 20.1 µmol, 1.2 equiv), sodium ascorbate (10.0 µL, 10.0 µmol, 1 M in H2O, 0.6 equiv) and copper(II) sulfate (8.3 µL, 8.3 µmol, 1 M in H2O, 0.5 equiv). After stirring for 16 h, the mixture was concentrated and the obtained crude product was subjected to reversed-phase flash chromatography (FlashPure Select C18, H2O + 0.1% HCOOH:MeCN 90:10–MeCN) and further purified by preparative HPLC (Luna C18, H2O + 0.1% HCOOH:MeCN 90:10–MeCN). Triazole 9g (12.4 mg, 13.3 µmol, 80%) was obtained as colorless powder after lyophilization. = –5.3 (c = 1.0, CHCl3). 1H-NMR (500 MHz, DMSO-D6): δ = 0.68 (d, J = 6.7 Hz, 3 H), 0.84 (d, J = 6.3 Hz, 3 H), 0.86–0.92 (m, 3 H), 0.94 (d, J = 6.7 Hz, 3 H), 1.16 (m, 1 H), 1.22–1.34 (m, 3 H), 1.36 (s, 9 H), 1.77 (m, 1 H), 2.48 (m, 1 H), 2.75–2.82 (m, J = 15.3, 11.7 Hz, 4 H), 2.92 (dd, J = 15.1, 4.9 Hz, 1 H), 2.95–3.03 (m, 2 H), 3.04 (dt, J = 6.1, 6.1 Hz, 2 H), 3.09 (dd, J = 15.1, 4.6 Hz, 1 H), 3.36 (t, J = 6.1 Hz, 2 H), 3.45–3.47 (m, 4 H), 3.48 (m, 2 H), 3.51 (m, 2 H), 3.57 (m, 1 H), 3.83 (ddd, J = 16.3, 10.8, 5.2 Hz, 1 H), 3.83 (ddd, J = 16.3, 11.1, 5.2 Hz, 1 H), 4.21 (d, J = 4.0 Hz, 1 H), 4.49–4.61 (m, 3 H), 5.22 (m, 1 H), 5.40 (dd, J = 11.7, 4.5 Hz, 1 H), 6.68–6.77 (m, 2 H), 6.98 (dd, J = 8.3, 1.9 Hz, 1 H), 7.47 (d, J = 1.8 Hz, 1 H), 7.91 (s, 1 H), 8.16 (dd, J = 8.7, 3.2 Hz, 1 H), 10.2 (bs, 1 H) ppm. 13C-NMR (125 MHz, DMSO-D6): δ = 14.4, 17.5, 18.6, 26.2, 28.2, 30.4, 30.8, 30.9, 31.3, 34.4, 37.1, 38.5, 39.7, 43.0, 44.7, 49.1, 56.5, 63.8, 69.0, 69.2, 69.5, 69.5, 69.6, 69.7, 70.7, 77.6, 84.3, 114.7, 123.2, 129.6, 130.0, 138.5, 141.9, 155.1, 155.6, 169.9, 170.7, 176.1 ppm. HRMS (ESI-ToF) calcd. for C40H64IN6O11+ [M+H]+: 931.3672 found 931.3672.
4.3.8. Synthesis of (3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-16-(3-phenylprop-2-yn-1-yl)-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (10a)
According to the general procedure for Sonogashira couplings, alkyne 8c (10.5 mg, 17.1 µmol) was reacted with (PPh3)2PdCl2 (1.2 mg, 1.71 µmol, 0.1 equiv), iodobenzene (17.5 mg, 85.7 µmol, 5.0 equiv) and copper(I) iodide (0.7 mg, 3.4 µmol, 0.2 equiv) in NEt3 (114 µL, 0.15 M) and stirred for 16 h. The obtained crude product was subjected to flash chromatography (RediSep Rf Silica, DCM–DCM:MeOH 90:10) and further purified by preparative HPLC (Luna C18, H2O:MeCN 90:10–MeCN). Alkyne 10a (6.8 mg, 9.9 µmol, 58%) was obtained as colorless powder after lyophilization. = –0.3 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.86 (d, J = 6.9 Hz, 3 H), 0.97 (d, J = 6.3 Hz, 3 H), 1.02–1.11 (m, 3 H), 1.13 (d, J = 6.7 Hz, 3 H), 1.20 (m, 1 H), 1.44 (ddd, J = 13.7, 11.4, 1.8 Hz, 1 H), 1.54 (m, 1 H), 1.60 (ddd, J = 13.7, 11.4 Hz, 1.8 Hz, 1 H), 2.02 (m, 1 H), 2.35 (bs, 1 H), 2.41 (ddq, J = 12.0, 6.6, 3.4 Hz, 1 H), 2.73 (d, J = 6.3 Hz, 1 H), 2.76 (dd, J = 15.6, 12.2 Hz, 1 H), 2.90 (s, 3 H), 3.25 (dd, J = 16.9, 1.7 Hz, 1 H), 3.35 (dd, J = 15.6, 4.6 Hz, 1 H), 3.66 (m, 1 H), 4.80 (dd, J = 16.8, 8.7 Hz, 1 H), 5.38 (ddt, J = 11.4, 6.4, 1.8 Hz, 1 H), 5.46 (dd, J = 12.1, 4.7 Hz, 1 H), 5.58 (bs, 1 H), 6.23 (dd, J = 8.7, 1.7 Hz, 1 H), 6.84 (d, J = 8.2 Hz, 1 H), 6.92 (dd, J = 8.3, 1.8 Hz, 1 H), 7.28 (d, J = 1.8 Hz, 1 H), 7.32–7.37 (m, 3 H), 7.43 (m, 2 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.3, 17.6, 18.4, 26.1, 27.1, 30.5, 32.6, 33.3, 34.5, 39.1, 39.7, 42.8, 45.0, 57.7, 65.9, 70.9, 82.8, 85.3, 85.7, 115.1, 123.1, 128.2, 128.5, 129.6, 130.4, 131.6, 137.8, 153.9, 171.2, 171.9, 177.5 ppm. HRMS (ESI-ToF) calcd. for C33H42IN2O6+ [M+H]+: 689.2062 found 689.2061.
4.3.9. Synthesis of (3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-16-[3-(4-methoxyphenyl)prop-2-yn-1-yl]-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (10b)
According to the general procedure for Sonogashira couplings, alkyne 8c (11.3 mg, 18.4 µmol) was reacted with (PPh3)2PdCl2 (1.3 mg, 1.85 µmol, 0.1 equiv), 1-iodo-4-methoxybenzene (21.6 mg, 92.3 µmol, 5.0 equiv) and copper(I) iodide (0.7 mg, 3.7 µmol, 0.2 equiv) in NEt3 (123 µL, 0.15 M) and stirred for 16 h. The obtained crude product was subjected to flash chromatography (RediSep Rf Silica, DCM–DCM:MeOH 9:1). Alkyne 10b (10.0 mg, 13.9 µmol, 75%) was obtained as colorless powder after lyophilization. = +8.8 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.85 (d, J = 6.9 Hz, 1 Hz, 3 H), 0.96 (d, J = 6.1 Hz, 3 H), 1.00–1.11 (m, 3 H), 1.13 (d, J = 6.6 Hz, 3 H), 1.19 (m, 1 H), 1.44 (m, 1 H), 1.54 (m, 1 H), 1.59 (m, 1 H), 2.01 (m, 1 H), 2.35–2.46 (m, J = 12.2, 6.6 Hz, 3.4 Hz, 1 H), 2.71 (d, J = 6.3 Hz, 2 H), 2.77 (dd, J = 15.6, 12.2 Hz, 1 H), 2.91 (s, 3 H), 3.23 (dd, J = 16.8, 1.8 Hz, 1 H), 3.35 (dd, J = 15.6, 4.6 Hz, 1 H), 3.66 (m, 1 H), 3.82 (s, 3 H), 4.80 (dd, J = 16.8, 8.7 Hz, 1 H), 5.36 (ddt, J = 11.4, 6.3, 2.0 Hz, 1 H), 5.47 (dd, J = 12.2, 4.6 Hz, 1 H), 6.12 (bs, 1 H), 6.29 (dd, J = 8.7, 1.8 Hz, 1 H), 6.83 (d, J = 8.4 Hz, 1 H), 6.87 (m, 2 H), 6.93 (dd, J = 8.4, 2.0 Hz, 1 H), 7.29 (d, J = 2.0 Hz, 1 H), 7.36 (m, 2 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.3, 17.6, 18.4, 26.1, 27.1, 30.5, 32.6, 33.2, 34.5, 39.1, 39.7, 42.8, 44.9, 55.3, 57.7, 65.9, 71.0, 82.6, 83.7, 85.4, 114.1, 115.1, 115.2, 129.5, 130.3, 133.0, 138.0, 154.1, 159.5, 171.1, 172.0, 177.5 ppm. HRMS (ESI-ToF) calcd. for C34H44IN2O7+ [M+H]+: 719.2188 found 719.2192.
4.3.10. Synthesis of (3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-16-[3-(4-nitrophenyl)prop-2-yn-1-yl]-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (10c)
According to the general procedure for Sonogashira couplings, alkyne 8c (14.4 mg, 23.5 µmol) was reacted with (PPh3)2PdCl2 (1.7 mg, 2.35 µmol, 0.1 equiv), 1-iodo-4-nitrobenzene (29.3 mg, 118 µmol, 5.0 equiv) and copper(I) iodide (0.9 mg, 4.7 µmol, 0.2 equiv) in NEt3 (157 µL, 0.15 M) and stirred for 16 h. The obtained crude product was subjected to flash chromatography (RediSep Rf Silica, DCM–DCM:MeOH 9:1). Alkyne 10c (13.1 mg, 17.9 µmol, 76%) was obtained as colorless powder after lyophilization. = +11.7 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.86 (d, J = 6.7 Hz, 3 H), 0.96 (d, J = 6.3 Hz, 3 H), 1.04–1.13 (m, 3 H), 1.14 (d, J = 6.6 Hz, 3 H), 1.21 (m, 1 H), 1.51–1.63 (m, 3 H), 1.99 (m, 1 H), 2.12 (bs, 1 H), 2.44 (ddq, J = 12.0, 6.6, 3.4 Hz, 1 H), 2.74–2.81 (m, 2 H), 2.87 (dd, J = 17.4, 5.3 Hz, 1 H), 2.92 (s, 3 H), 3.17 (dd, J = 16.6, 2.1 Hz, 1 H), 3.39 (dd, J = 15.3, 4.6 Hz, 1 H), 3.72 (m, 1 H), 4.78 (dd, J = 16.6, 8.8 Hz, 1 H), 5.38 (m, 1 H), 5.56 (dd, J = 11.9, 4.7 Hz, 1 H), 6.36 (dd, J = 8.8, 2.1 Hz, 1 H), 6.70–6.91 (m, J = 8.2 Hz, 2 H), 6.99 (dd, J = 8.3, 2.0 Hz, 1 H), 7.37 (d, J = 2.0 Hz, 1 H), 7.62 (m, 2 H), 8.20 (m, 2 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.3, 17.6, 18.5, 26.0, 27.1, 30.7, 32.5, 32.7, 34.8, 39.1, 39.5, 42.9, 44.9, 57.6, 66.0, 70.3, 81.6, 85.2, 90.9, 115.1, 123.7, 129.5, 130.0, 130.0, 132.5, 138.2, 147.0, 154.3, 170.5, 172.0, 177.6 ppm. HRMS (ESI-ToF) calcd. for C33H41IN3O8+ [M+H]+: 734.1933 found 734.1936.
4.3.11. Synthesis of (3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-16-{3-[(2-hydroxyethyl)thio]propyl}-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (11a)
According to the general procedure for thiol-ene click reactions, alkene 8b (17.6 mg, 28.6 µmol) was dissolved in anhydrous THF (286 µL, 0.1 M) and reacted with 2-mercaptoethanol (5.00 µL, 57.3 µmol, 1.12 gml−1, 2.0 equiv), triethylborane (34.4 µL, 8.59 µmol, 0.3 equiv, 0.25 M in hexane) and air (0.4 mL) and stirred for 20 h. The obtained crude product was subjected to reversed-phase chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN). Thioether 11a (14.2 mg, 20.5 µmol, 72%) was obtained as colorless powder after lyophilization. = –14.9 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.83 (d, J = 6.7 Hz, 3 H), 0.94 (d, J = 6.0 Hz, 3 H), 1.01–1.10 (m, 3 H), 1.12 (d, J = 6.6 Hz, 3 H), 1.17 (m, 1 H), 1.35 (ddd, J = 13.9, 11.5, 2.0 Hz, 1 H), 1.45 (ddd, J = 13.8, 11.6, 2.0 Hz, 1 H), 1.51 (m, 1 H), 1.62 (m, 2 H), 1.72 (m, 2 H), 1.97 (m, 1 H), 2.14–2.35 (m, 2 H), 2.43 (ddq, J = 11.7, 6.6, 3.4 Hz, 1 H), 2.55 (m, 2 H), 2.73 (m, 2 H), 2.86 (dd, J = 15.3, 12.1 Hz, 1 H), 2.93 (s, 3 H), 3.15 (dd, J = 16.7, 2.0 Hz, 1 H), 3.40 (dd, J = 15.3, 4.4 Hz, 1 H), 3.63 (ddd, J = 11.6, J = 4.0, 2.0 Hz, 1 H), 3.74 (t, J = 6.0 Hz, 2 H), 4.76 (dd, J = 16.7, 8.8 Hz, 1 H), 5.22 (m, 1 H), 5.49 (dd, J = 12.1, 4.4 Hz, 1 H), 6.40 (dd, J = 8.8, 2.0 Hz, 1 H), 6.83 (d, J = 8.2 Hz, 1 H), 7.04 (dd, J = 8.2, 1.8 Hz, 1 H), 7.48 (d, J = 1.8 Hz, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.4, 17.6, 18.4, 25.6, 27.0, 30.9, 31.3, 32.6, 33.1, 34.0, 34.4 35.1, 39.1, 39.5, 43.0, 44.8, 58.0, 60.7, 66.0, 72.4 85.2, 115.2, 129.6, 130.2, 138.3, 154.3, 171.0, 171.9, 177.9 ppm. HRMS (ESI-ToF) calcd. for C29H46IN2O7+ [M+H]+: 693.2065 found 693.2076.
4.3.12. Synthesis of (3R,9S,11S,13R,14S,16R)-16-[3-(Butylthio)propyl]-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (11b)
According to the general procedure for thiol-ene click reactions, alkene 8b (17.9 mg, 29.1 µmol) was dissolved in anhydrous THF (291 µL, 0.1 M) and reacted with 1-butanethiol (6.25 µL, 58.2 µmol, 0.84 gml−1, 2.0 equiv), triethylborane (35.0 µL, 8.74 µmol, 0.3 equiv, 0.25 M in hexane) and air (0.4 mL) and stirred for 20 h. The obtained crude product was subjected to reversed-phase chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN). Thioether 11b (17.0 mg, 24.1 µmol, 83%) was obtained as colorless powder after lyophilization. = −13.2 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.83 (d, J = 6.9 Hz, 3 H), 0.90–0.97 (m, J = 6.7 Hz, 6 H), 1.01–1.10 (m, 3 H), 1.12 (d, J = 6.7 Hz, 3 H), 1.19 (m, 1 H), 1.32 (m, 1 H), 1.41 (m, 2 H), 1.45–1.53 (m, 2 H), 1.54–1.63 (m), 1.67 (m, 1 H), 1.76 (m, 1 H), 1.98 (m, 1 H), 2.44 (ddq, J = 11.6, 6.7, 3.2 Hz, 1 H), 2.46–2.58 (m, 4 H), 2.86 (dd, J = 15.3, 12.3 Hz, 1 H), 2.92 (s, 3 H), 3.22 (d, J = 16.7 Hz, 1 H), 3.41 (dd, J = 15.4, 4.4 Hz, 1 H), 3.62 (ddd, J = 11.4, 4.0, 2.0 Hz, 1 H), 4.77 (dd, J = 16.7, 8.6 Hz, 1 H), 5.20 (m, 1 H), 5.49 (dd, J = 12.3, 4.4 Hz, 1 H), 6.34 (m, 1 H), 6.83 (m, 1 H), 7.05 (dd, J = 8.3, 2.0 Hz, 1 H), 7.48 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 13.7, 14.4, 17.6, 18.4, 22.0, 25.8, 27.0, 30.9, 31.7, 31.8, 32.0, 32.7, 33.5, 34.4, 34.4, 39.1, 39.6, 43.0, 44.9, 58.0, 65.9, 72.6, 85.3, 115.2, 129.5, 130.2, 138.2, 154.3, 171.1, 172.0, 177.7 ppm. HRMS (ESI-ToF) calcd. for C31H50IN2O6S+ [M+H]+: 705.2429 found 705.2426.
4.3.13. Synthesis of (3R,9S,11S,13R,14S,16R)-16-[3-(Benzylthio)propyl]-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-1-oxa-4,7-diazacyclohexadecane-2,5,8-trione (11c)
According to the general procedure for thiol-ene click reactions, alkene 8b (18.8 mg, 30.6 µmol) was dissolved in anhydrous THF (306 µL, 0.1 M) and reacted with benzyl mercaptan (7.17 µL, 61.2 µmol, 1.06 gml−1, 2.0 equiv), triethylborane (36.7 µL, 9.18 µmol, 0.3 equiv, 0.25 M in hexane) and air (0.4 mL) and stirred for 20 h. The obtained crude product was subjected to reversed-phase chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN). Thioether 11c (12.5 mg, 16.9 µmol, 55%) was obtained as colorless powder after lyophilization. = –6.7 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.83 (d, J = 6.7 Hz, 3 H), 0.95 (d, J = 6.1 Hz, 3 H), 1.02–1.09 (m, 3 H), 1.13 (d, J = 6.7 Hz, 3 H), 1.17 (m, 1 H), 1.29 (m, 1 H), 1.43 (m, 1 H), 1.48–1.59 (m, 3 H), 1.63 (m, 1 H), 1.71 (m, 1 H), 1.98 (m, 1 H), 2.39–2.47 (m, 3 H), 2.84 (dd, J = 15.4, 12.2 Hz, 1 H), 2.88 (s, 3 H), 3.22 (d, J = 16.6 Hz, 1 H), 3.38 (dd, J = 15.4, 4.4 Hz, 1 H), 3.60 (ddd, J = 11.4, 4.2, 2.0 Hz, 1 H), 3.71 (s, 2 H), 4.76 (dd, J = 16.6, 8.7 Hz, 1 H), 5.16 (m, 1 H), 5.46 (dd, J = 12.2, 4.4 Hz, 1 H), 6.29 (d, J = 8.7 Hz, 1 H), 6.85 (d, J = 8.2 Hz, 1 H), 7.04 (dd, J = 8.2, 2.0 Hz, 1 H), 7.24 (m, 1 H), 7.29–7.34 (m, 4 H), 7.47 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.4, 17.6, 18.4, 25.4, 27.0, 30.8, 31.0, 32.6, 33.5, 34.4, 34.4, 36.5, 39.1, 39.6, 43.0, 44.9, 58.0, 65.8, 72.6, 85.4, 115.2, 127.0, 128.5, 128.8, 129.6, 130.3, 138.1, 138.4, 154.2, 171.2, 171.9, 177.7 ppm. HRMS (ESI-ToF) calcd. for C34H48IN2O6S+ [M+H]+: 739.2272 found 739.2298.
4.3.14. Synthesis of Ethyl 3-({3-[(3R,9S,11S,13R,14S,16R)-14-hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-2,5,8-trioxo-1-oxa-4,7-diazacyclohexadecan-16-yl]propyl}-thio)propanoate (11d)
According to the general procedure for thiol-ene click reactions, alkene 8b (18.8 mg, 30.6 µmol) was dissolved in anhydrous THF (306 µL, 0.1 M) and reacted with ethyl 3-mercaptopropanoate (7.75 µL, 61.2 µmol, 1.06 gml−1, 2.0 equiv), triethylborane (36.7 µL, 9.18 µmol, 0.3 equiv, 0.25 M in hexane) and air (0.4 mL). After stirring for 2 h, triethylborane (36.7 µL, 9.18 µmol, 0.3 equiv, 0.25 M in hexane) was added and stirred for another 24 h. The obtained crude product was subjected to reversed-phase chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN) and further purified by preparative HPLC (Luna C18, H2O:MeCN 25:75–MeCN). Thioether 11d (19.2 mg, 25.6 µmol, 79%) was obtained as colorless powder after lyophilization. = –8.5 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.83 (d, J = 6.7 Hz, 3 H), 0.95 (d, J = 6.0 Hz, 3 H), 1.00–1.09 (m, 3 H), 1.12 (d, J = 6.6 Hz, 3 H), 1.19 (m, 1 H), 1.27 (t, J = 7.2 Hz, 3 H), 1.33 (m, 1 H), 1.43–1.55 (m, 2 H), 1.56–1.71 (m, 3 H), 1.76 (m, 1 H), 1.85 (bs, 1 H), 1.98 (m, 1 H), 2.43 (m, 1 H), 2.55 (m, 2 H), 2.60 (t, J = 7.4 Hz, 2 H), 2.78 (t, J = 7.4 Hz, 2 H), 2.88 (dd, J = 15.3, 11.9 Hz, 1 H), 2.93 (s, 3 H), 3.22 (d, J = 16.5 Hz, 1 H), 3.41 (dd, J = 15.3, 4.0 Hz, 1 H), 3.62 (m, 1 H), 4.16 (q, J = 7.2 Hz, 2 H), 4.77 (dd, J = 16.5, 7.7 Hz, 1 H), 5.21 (m, 1 H), 5.48 (dd, J = 12.0, 4.2 Hz, 1 H), 6.33 (bs, 1 H), 6.84 (d, J = 8.2 Hz, 1 H), 7.06 (d, J = 7.9 Hz, 1 H), 7.50 (s, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.2, 14.4, 17.6, 18.4, 25.6, 27.0, 27.0, 30.9, 31.7, 32.6, 33.5, 34.3, 34.4, 34.9, 39.1, 39.6, 43.0, 44.9, 58.1, 60.7, 65.9, 72.5, 85.2, 115.2, 129.6, 130.3, 138.2, 154.3, 171.2, 171.9, 172.0, 177.7 ppm. HRMS (ESI-ToF) calcd. for C32H50IN2O8S+ [M+H]+: 749.2327 found 749.2360.
4.3.15. Synthesis of 2-({3-[(3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-2,5,8-trioxo-1-oxa-4,7-diazacyclohexadecan-16-yl]propyl}-thio)acetic Acid (11e)
According to the general procedure for thiol-ene click reactions, alkene 8b (19.1 mg, 31.1 µmol) was dissolved in anhydrous THF (296 µL, 0.1 M) and reacted with 2-mercaptoacetic acid (4.31 µL, 62.2 µmol, 1.33 gml−1, 2.0 equiv), triethylborane (37.3 µL, 9.32 µmol, 0.3 equiv, 0.25 M in hexane) and air (0.4 mL). After stirring for 2 h, triethylborane (37.3 µL, 9.32 µmol, 0.3 equiv, 0.25 M in hexane) was added and stirred for another 18 h. The obtained crude product was subjected to reversed-phase chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN) and further purified by preparative HPLC (Luna C18, H2O:MeCN 25:75–MeCN). Thioether 11e (14.4 mg, 20.4 µmol, 66%) was obtained as colorless powder after lyophilization. = −11.5 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.84 (d, J = 6.7 Hz, 3 H), 0.94 (d, J = 6.0 Hz, 3 H), 1.03–1.11 (m, 3 H), 1.11–1.18 (m, J = 6.6 Hz, 4 H), 1.36 (m, 1 H), 1.44 (m, 1 H), 1.56 (m, 1 H), 1.60–1.77 (m, 3 H), 1.82 (m, 1 H), 1.99 (m, 1 H), 2.46 (ddq, J = 12.1, 6.6, 3.4 Hz, 1 H), 2.62 (m, 2 H), 2.86 (dd, J = 15.5, 12.1 Hz, 1 H), 2.98 (s, 3 H), 3.16 (d, J = 16.8 Hz, 1 H), 3.23 (s, 2 H), 3.40 (dd, J = 15.4, 4.4 Hz, 1 H), 3.65 (ddd, J = 11.3, 4.1, 2.1 Hz, 1 H), 4.96 (dd, J = 16.8, 9.0 Hz, 1 H), 5.27 (m, 1 H), 5.55 (dd, J = 12.1, 4.4 Hz, 1 H), 6.72 (bs, 1 H), 6.84 (d, J = 8.2 Hz, 1 H), 7.04 (d, J = 8.4, 1.8 Hz, 1 H), 7.48 (d, J = 2.0 Hz, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.2, 17.5, 18.3, 25.0, 26.9, 30.9, 31.9, 32.4, 33.4, 33.5, 34.3, 34.6, 39.2, 39.7, 42.9, 44.6, 57.7, 66.0, 72.1, 85.3, 115.2, 129.6, 130.3, 138.2, 154.3, 170.8, 171.8, 173.4, 178.5 ppm. HRMS (ESI-ToF) calcd. for C29H44IN2O6S+ [M+H]+: 707.1858 found 707.1884.
4.3.16. Synthesis of S-{3-[(3R,9S,11S,13R,14S,16R)-14-Hydroxy-3-(4-hydroxy-3-iodobenzyl)-4,9,11,13-tetramethyl-2,5,8-trioxo-1-oxa-4,7-diazacyclohexadecan-16-yl]propyl} ethanethioate (11f)
According to the general procedure for thiol-ene click reactions, alkene 8b (18.0 mg, 29.3 µmol) was dissolved in anhydrous THF (293 µL, 0.1 M) and reacted with thioacetic acid (4.17 µL, 58.6 µmol, 1.07 gml−1, 2.0 equiv), triethylborane (35.1 µL, 8.79 µmol, 0.3 equiv, 0.25 M in hexane) and air (0.4 mL) and stirred for 20 h. The obtained crude product was subjected to reversed-phase chromatography (FlashPure Select C18, H2O:MeCN 90:10–MeCN). Thioether 11f (19.0 mg, 27.5 µmol, 94%) was obtained as colorless powder after lyophilization. = −11.6 (c = 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3): δ = 0.83 (d, J = 6.7 Hz, 3 H), 0.95 (d, J = 6.1 Hz, 3 H), 1.00–1.09 (m, 3 H), 1.12 (d, J = 6.6 Hz, 3 H), 1.18 (m, 1 H), 1.32 (m, 1 H), 1.45 (m, 1 H), 1.52 (m, 1 H), 1.56–1.66 (m, 3 H), 1.72 (m, 1 H), 1.99 (m, 1 H), 2.34 (s, 3 H), 2.44 (m, 1 H), 2.85 (t, J = 6.8 Hz, 2 H), 2.89 (m, 1 H), 2.93 (s, 3 H), 3.22 (d, J = 16.6 Hz, 1 H), 3.40 (dd, J = 15.4, 4.4 Hz, 1 H), 3.61 (ddd, J = 11.3, 4.0, 1.9 Hz, 1 H), 4.77 (dd, J = 16.8, 8.7 Hz, 1 H), 5.20 (m, 1 H), 5.48 (dd, J = 12.1, 4.3 Hz, 1 H), 6.33 (bs, 1 H), 6.74 (bs, 1 H), 6.83 (d, J = 8.4 Hz, 1 H), 7.06 (dd, J = 8.4, 1.8 Hz, 1 H), 7.49 (d, J = 1.8 Hz, 1 H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 14.4, 17.6, 18.4, 25.9, 27.0, 28.7, 30.7, 30.9, 32.6, 33.6, 34.3, 34.4, 39.1, 39.6, 43.0, 44.9, 58.1, 65.8, 72.4, 85.3, 115.2, 129.6, 130.3, 138.2, 154.2, 171.2, 171.9, 177.7, 195.9 ppm. HRMS (ESI-ToF) calcd. for C29H44IN2O7S+ [M+H]+: 691.1908 found 691.1909.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}