
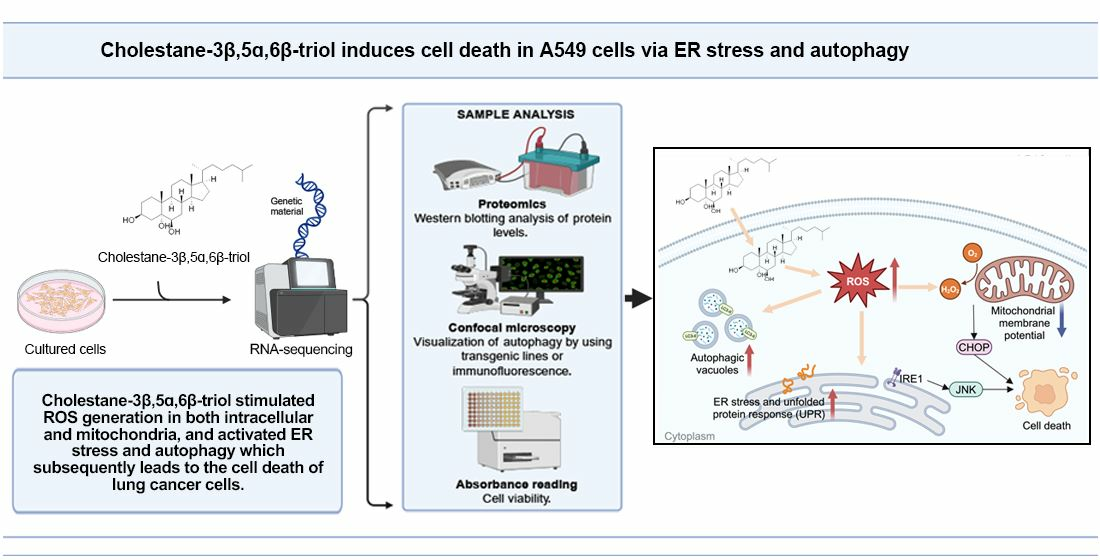
Cholestane-3β,5α,6β-triol Induces Multiple Cell Death in A549 Cells via ER Stress and Autophagy Activation

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
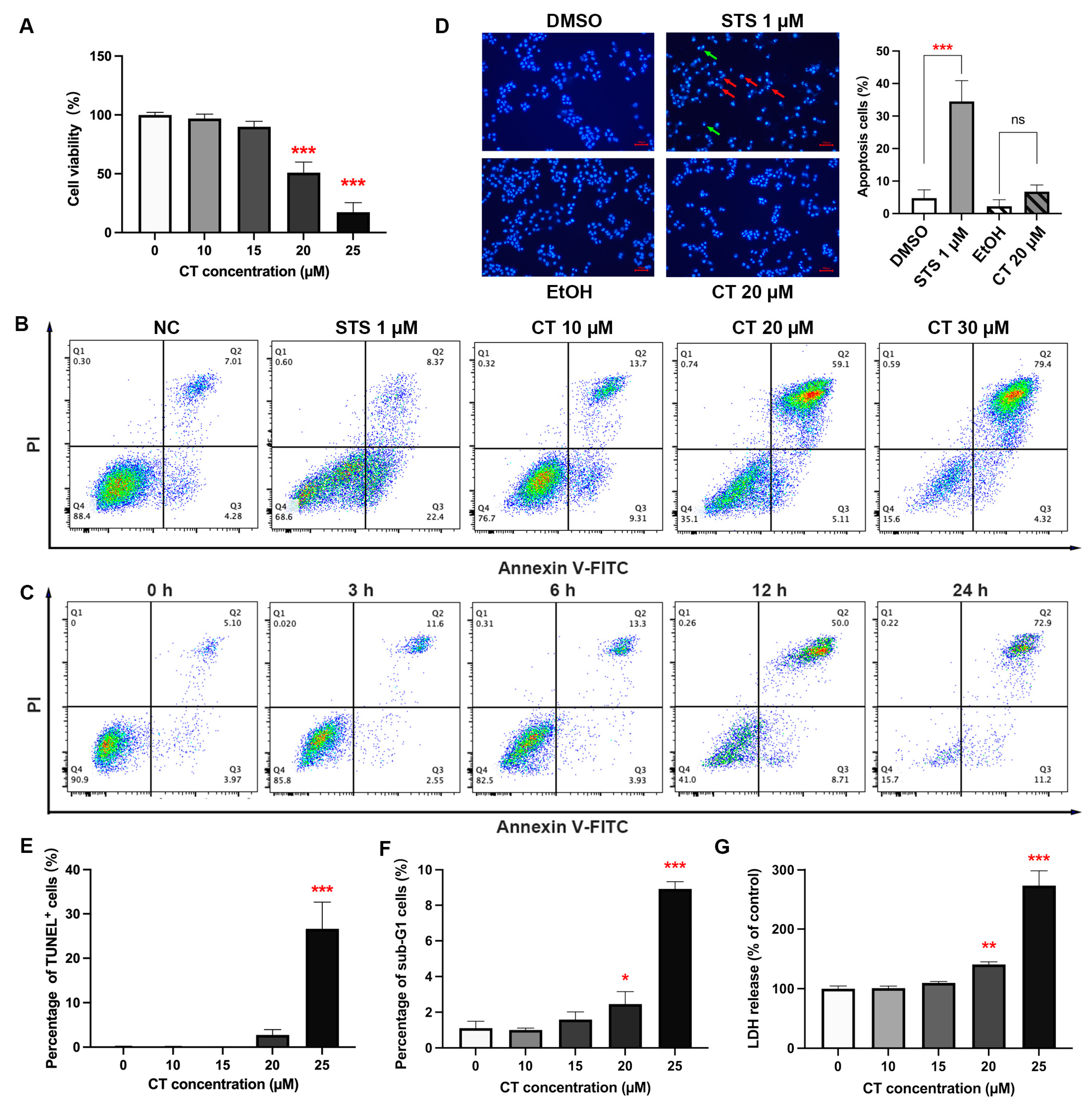
2.1. Effect of CT on A549 Cell Death
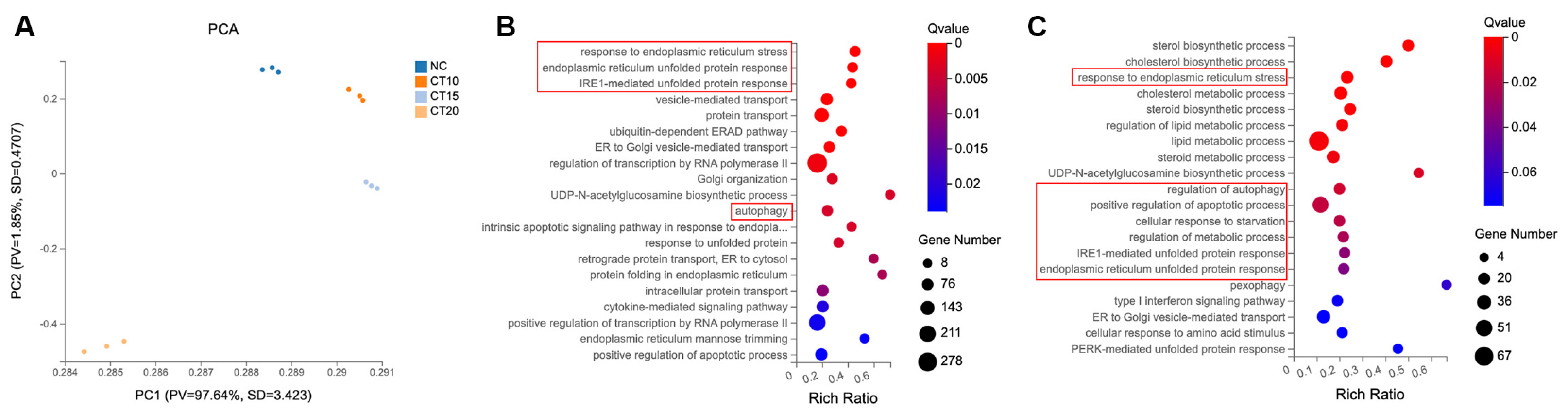
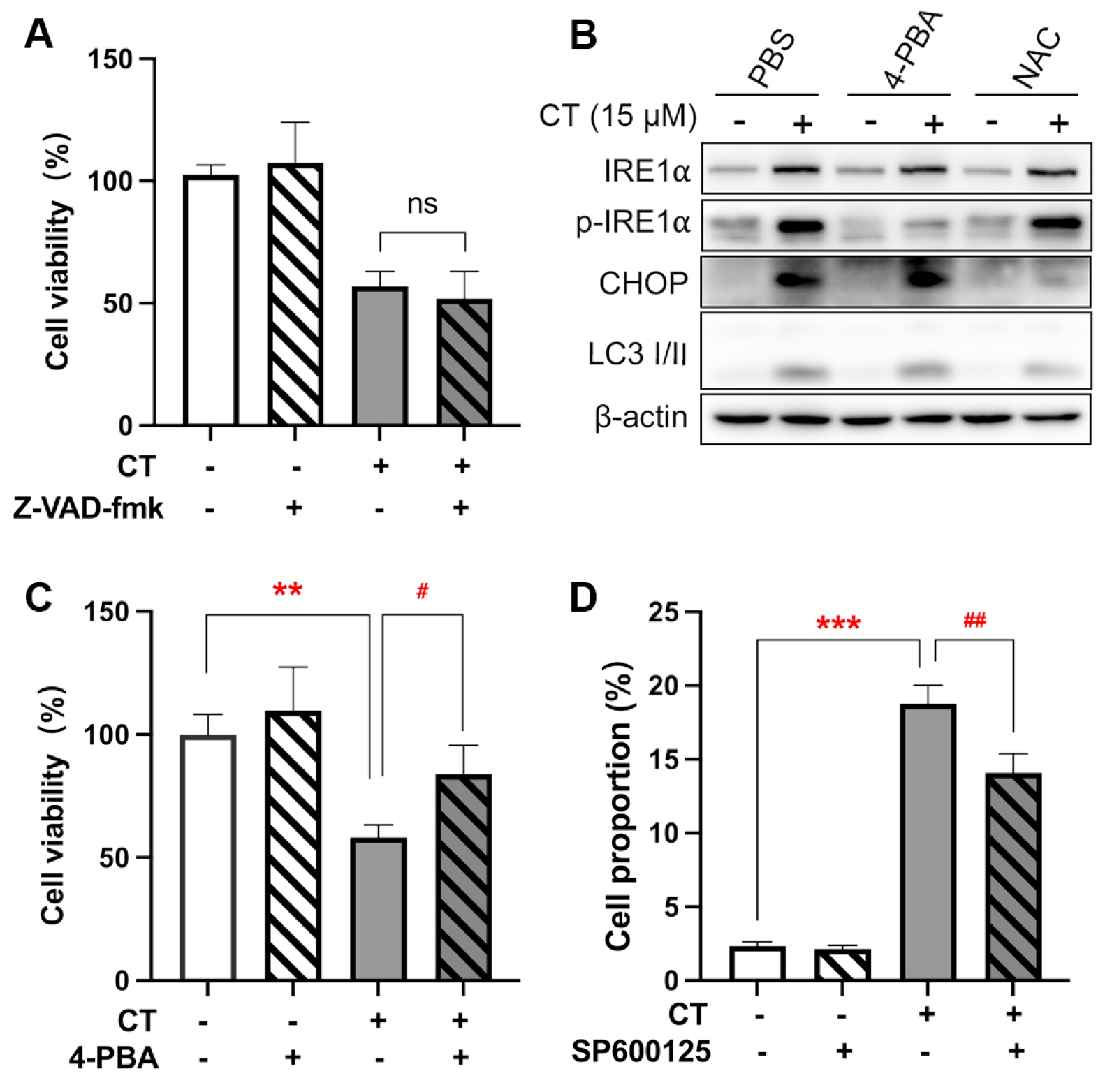
2.2. CT Activated the Response to ER Stress in A549 Cells
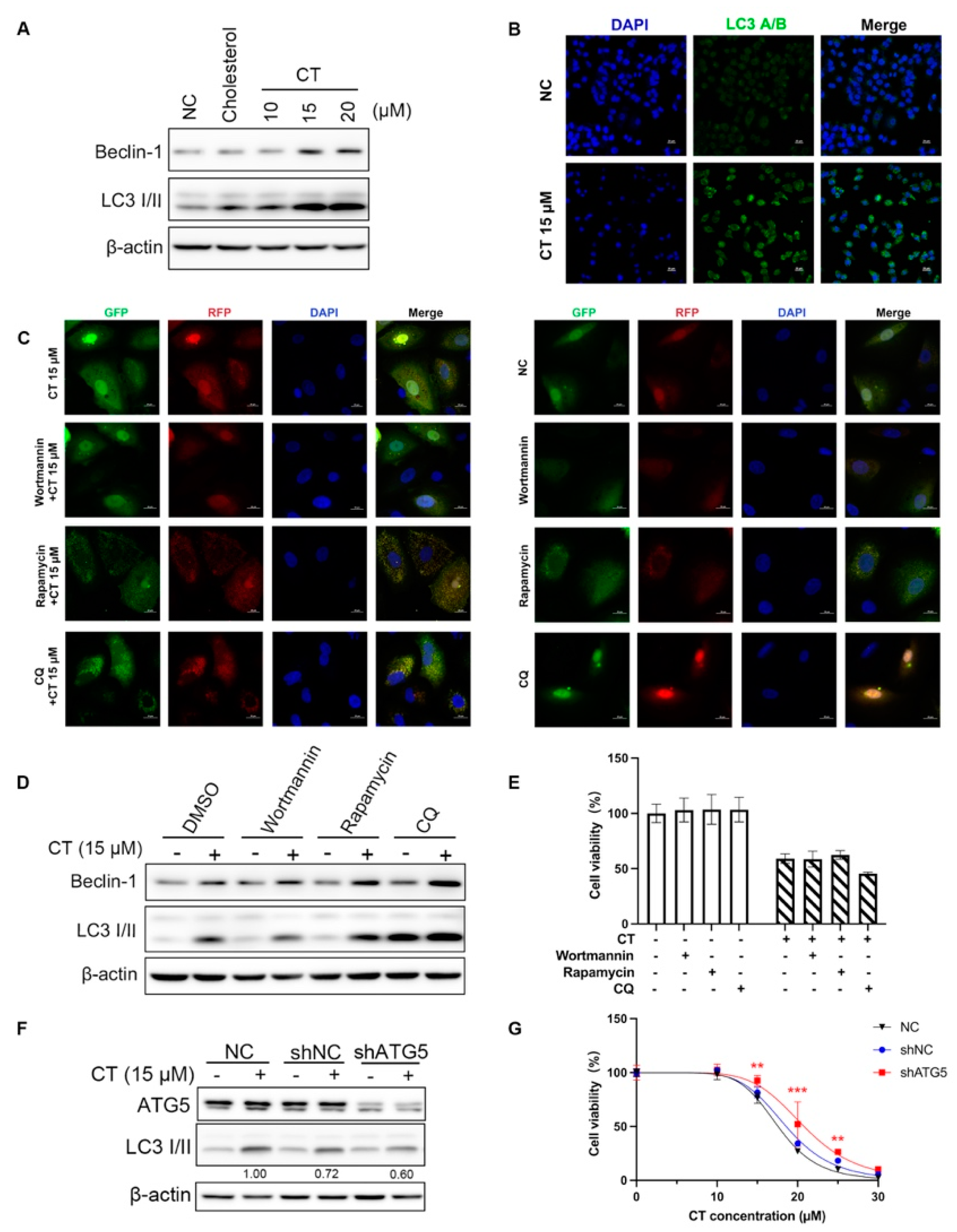
2.3. CT Enhanced Autophagy Flux in A549 Cells
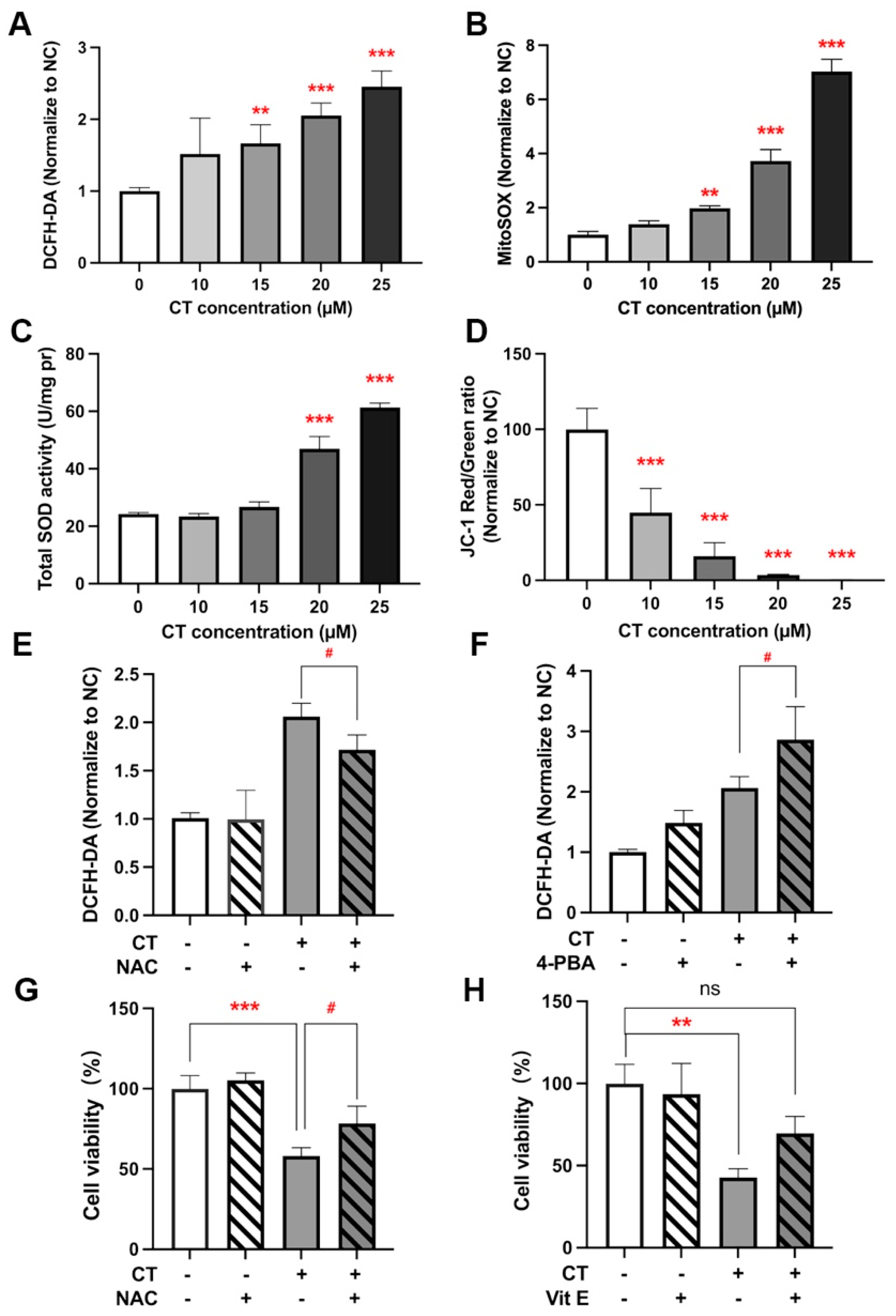
2.4. CT-Induced ROS Generation Triggered ER Stress and Autophagy
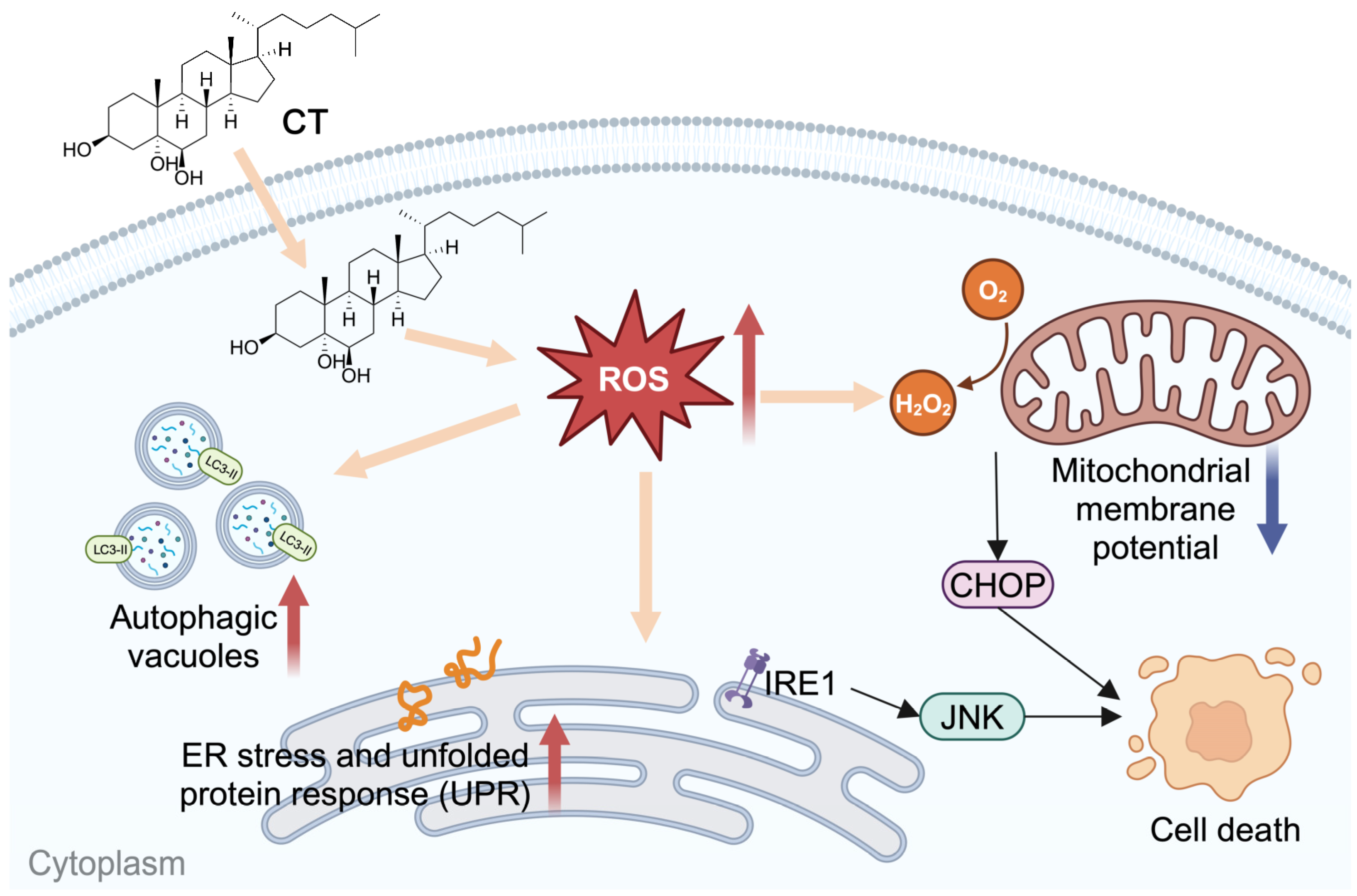
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Savić, M.P.; Sakač, M.N.; Kuzminac, I.Z.; Ajduković, J.J. Structural diversity of bioactive steroid compounds isolated from soft corals in the period 2015–2020. J. Steroid Biochem. Mol. Biol. 2022, 218, 106061. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2023, 40, 275–325. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, H.; Wang, P.; Gong, W.; Xue, M.; Zhang, H.; Liu, T.; Liu, B.; Yi, Y.; Zhang, W. Bioactive polyoxygenated steroids from the South China sea soft coral, Sarcophyton sp. Mar. Drugs 2013, 11, 775–787. [Google Scholar] [CrossRef]
- Liu, T.-F.; Lu, X.; Tang, H.; Zhang, M.-M.; Wang, P.; Sun, P.; Liu, Z.-Y.; Wang, Z.-L.; Li, L.; Rui, Y.-C.; et al. 3β,5α,6β-Oxygenated sterols from the South China Sea gorgonian Muriceopsis flavida and their tumor cell growth inhibitory activity and apoptosis-inducing function. Steroids 2013, 78, 108–114. [Google Scholar] [CrossRef]
- Wang, P.; Tang, H.; Liu, B.S.; Li, T.J.; Sun, P.; Zhu, W.; Luo, Y.P.; Zhang, W. Tumor cell growth inhibitory activity and structure-activity relationship of polyoxygenated steroids from the gorgonian Menella kanisa. Steroids 2013, 78, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, J.; Su, L.; Tang, H.; Zhang, W. Osteoclastogenesis modulatory steroids from the South China Sea gorgonian coral Iciligorgia sp. Chem. Biodivers. 2020, 17, e2000266. [Google Scholar] [CrossRef] [PubMed]
- Luoma, P.V. Cytochrome P450—physiological key factor against cholesterol accumulation and the atherosclerotic vascular process. Ann. Med. 2007, 39, 359–370. [Google Scholar] [CrossRef]
- Iuliano, L. Pathways of cholesterol oxidation via non-enzymatic mechanisms. Chem. Phys. Lipids 2011, 164, 457–468. [Google Scholar] [CrossRef] [PubMed]
- de Medina, P.; Paillasse, M.R.; Segala, G.; Poirot, M.; Silvente-Poirot, S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 13520–13525. [Google Scholar] [CrossRef]
- Zmysłowski, A.; Szterk, A. Oxysterols as a biomarker in diseases. Clin. Chim. Acta 2019, 491, 103–113. [Google Scholar] [CrossRef]
- Samadi, A.; Sabuncuoglu, S.; Samadi, M.; Isikhan, S.Y.; Chirumbolo, S.; Peana, M.; Lay, I.; Yalcinkaya, A.; Bjørklund, G. A Comprehensive review on oxysterols and related diseases. Curr. Med. Chem. 2021, 28, 110–136. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, F.A.; Levy, D.; Zarrouk, A.; Lizard, G.; Bydlowski, S.P. Impact of oxysterols on cell death, proliferation, and differentiation induction: Current status. Cells 2021, 10, 2301. [Google Scholar] [CrossRef] [PubMed]
- Nury, T.; Yammine, A.; Ghzaiel, I.; Sassi, K.; Zarrouk, A.; Brahmi, F.; Samadi, M.; Rup-Jacques, S.; Vervandier-Fasseur, D.; Pais de Barros, J.P.; et al. Attenuation of 7-ketocholesterol- and 7β-hydroxycholesterol-induced oxiapoptophagy by nutrients, synthetic molecules and oils: Potential for the prevention of age-related diseases. Ageing Res. Rev. 2021, 68, 101324. [Google Scholar] [CrossRef]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C.; et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef] [PubMed]
- Prunet, C.; Lemaire-Ewing, S.; Ménétrier, F.; Néel, D.; Lizard, G. Activation of caspase-3-dependent and -independent pathways during 7-ketocholesterol- and 7β-hydroxycholesterol-induced cell death: A morphological and biochemical study. J. Biochem. Mol. Toxicol. 2005, 19, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Monier, S.; Samadi, M.; Prunet, C.; Denance, M.; Laubriet, A.; Athias, A.; Berthier, A.; Steinmetz, E.; Jürgens, G.; Nègre-Salvayre, A.; et al. Impairment of the cytotoxic and oxidative activities of 7β-hydroxycholesterol and 7-ketocholesterol by esterification with oleate. Biochem. Biophys. Res. Commun. 2003, 303, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Seye, C.I.; Knaapen, M.W.M.; Daret, D.; Desgranges, C.; Herman, A.G.; Kockx, M.A.; Bult, H. 7-ketocholesterol induces reversible cytochrome c release in smooth muscle cells in absence of mitochondrial swelling. Cardiovasc. Res. 2004, 64, 144–153. [Google Scholar] [CrossRef]
- Helmschrodt, C.; Becker, S.; Schröter, J.; Hecht, M.; Aust, G.; Thiery, J.; Ceglarek, U. Fast LC–MS/MS analysis of free oxysterols derived from reactive oxygen species in human plasma and carotid plaque. Clin. Chim. Acta 2013, 425, 3–8. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Kang, J.J.; Shih, Y.L.; Lo, Y.L.; Wang, C.F. Cholesterol-3β,5α,6β-triol induced genotoxicity through reactive oxygen species formation. Food Chem. Toxicol. 2005, 43, 617–622. [Google Scholar] [CrossRef]
- Liao, P.L.; Cheng, Y.W.; Li, C.H.; Lo, Y.L.; Kang, J.J. Cholesterol-3β,5α,6β-triol induced PI(3)K-Akt-eNOS-dependent cyclooxygenase-2 expression in endothelial cells. Toxicol. Lett. 2009, 190, 172–178. [Google Scholar] [CrossRef]
- Attanzio, A.; Frazzitta, A.; Cilla, A.; Livrea, M.A.; Tesoriere, L.; Allegra, M. 7-Ketocholesterol and cholestane-3β,5α,6β-Triol induce eryptosis through distinct pathways leading to NADPH oxidase and nitric oxide synthase activation. Cell Physiol. Biochem. 2019, 53, 933–947. [Google Scholar] [PubMed]
- Liu, H.; Wang, T.; Huang, K. Cholestane-3β,5α,6β-triol-induced reactive oxygen species production promotes mitochondrial dysfunction in isolated mice liver mitochondria. Chem. Biol. Interact. 2009, 179, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yuan, L.; Xu, S.; Wang, K.; Zhang, T. Cholestane-3β,5α,6β-triol inhibits osteoblastic differentiation and promotes apoptosis of rat bone marrow stromal cells. J. Cell Biochem. 2005, 96, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Yuan, L.; Xu, S.J.; Zhang, T.L.; Wang, K. Cholestane-3β,5α,6β-triol promotes vascular smooth muscle cells calcification. Life Sci. 2004, 76, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.F.S.M.M.; Moreira, J.N.; Joao, N.; Simoes, S.; Luisa Sa e Melo, M. Sterols as anticancer agents: Synthesis of ring-B oxygenated steroids, cytotoxic profile, and comprehensive SAR analysis. J. Med. Chem. 2010, 53, 7632–7638. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, C.; Huang, K. Lanthanum chloride suppresses oxysterol-induced ECV-304 cell apoptosis via inhibition of intracellular Ca2+ concentration elevation, oxidative stress, and activation of ERK and NF-κB signaling pathways. J. Biol. Inorg. Chem. 2011, 16, 671–681. [Google Scholar] [CrossRef]
- Levy, D.; Correa de Melo, T.; Ohira, B.Y.; Fidelis, M.L.; Ruiz, J.L.M.; Rodrigues, A.; Bydlowski, S.P. Oxysterols selectively promote short-term apoptosis in tumor cell lines. Biochem. Biophys. Res. Commun. 2018, 505, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Stein, D.; Slobodnik, Z.; Tam, B.; Einav, M.; Akabayov, B.; Berstein, S.; Toiber, D. 4-Phenylbutyric acid—Identity crisis; can it act as a translation inhibitor? Aging Cell 2022, 21, e13738. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Liu, L.; Han, C.; Yu, H.; Zhu, W.; Cui, H.; Zheng, L.; Zhang, C.; Yue, L. Chloroquine inhibits cell growth in human A549 lung cancer cells by blocking autophagy and inducing mitochondrial-mediated apoptosis. Oncol. Rep. 2018, 39, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. p62 at the Crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef]
- Ye, X.; Zhou, X.J.; Zhang, H. Exploring the role of autophagy-related gene 5 (ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fanto, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol. 2016, 10, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kreilaus, F.; Spiro, A.S.; McLean, C.A.; Garner, B.; Jenner, A.M. Evidence for altered cholesterol metabolism in Huntington’s disease post mortem brain tissue. Neuropathol. Appl. Neurobiol. 2016, 42, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, L.; Micheletta, F.; Natoli, S.; Ginanni Corradini, S.; Iappelli, M.; Elisei, W.; Giovannelli, L.; Violi, F.; Diczfalusy, U. Measurement of oxysterols and α-tocopherol in plasma and tissue samples as indices of oxidant stress status. Anal. Biochem. 2003, 312, 217–223. [Google Scholar] [CrossRef]
- Raselli, T.; Hearn, T.; Wyss, A.; Atrott, K.; Peter, A.; Frey-Wagner, I.; Spalinger, M.R.; Maggio, E.M.; Sailer, A.W.; Schmitt, J.; et al. Elevated oxysterol levels in human and mouse livers reflect nonalcoholic steatohepatitis. J. Lipid Res. 2019, 60, 1270–1283. [Google Scholar] [CrossRef]
- Linseisen, J.; Wolfram, G.N.; Miller, A.B. Plasma 7β-hydroxycholesterol as a possible predictor of lung cancer risk. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1630–1637. [Google Scholar]
- Kloudova-Spalenkova, A.; Ueng, Y.-F.; Wei, S.; Kopeckova, K.; Guengerich, F.P.; Soucek, P. Plasma oxysterol levels in luminal subtype breast cancer patients are associated with clinical data. J. Steroid Biochem. Mol. Biol. 2020, 197, 105566. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Martin, C.W.; Wynder, E.L. Fecal bile acids and cholesterol metabolites of patients with ulcerative colitis, a high-risk group for development of colon cancer. Cancer Res. 1977, 37, 1697–1701. [Google Scholar]
- Lin, C.Y.; Huo, C.; Kuo, L.K.; Hiipakka, R.A.; Jones, R.B.; Lin, H.P.; Hung, Y.; Su, L.C.; Tseng, J.C.; Kuo, Y.Y.; et al. Cholestane-3β,5α,6β-triol suppresses proliferation, migration, and invasion of human prostate cancer cells. PLoS ONE 2013, 8, e65734. [Google Scholar] [CrossRef]
- Jusakul, A.; Loilome, W.; Namwat, N.; Haigh, W.G.; Kuver, R.; Dechakhamphu, S.; Sukontawarin, P.; Pinlaor, S.; Lee, S.P.; Yongvanit, P. Liver fluke-induced hepatic oxysterols stimulate DNA damage and apoptosis in cultured human cholangiocytes. Mutat. Res. 2012, 731, 48–57. [Google Scholar] [CrossRef]
- Wu, Q.Z.; Huang, K.X. Protective effect of ebselen on cytotoxicity induced by cholestane-3β,5α,6β-triol in ECV-304 cells. Biochim. Biophys. Acta 2006, 1761, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, J.; Yang, N.; Huang, Y.; Hu, T.; Rao, C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022, 13, 1051. [Google Scholar] [CrossRef]
- Han, J.; Cheng, C.; Zhang, J.; Fang, J.; Yao, W.; Zhu, Y.; Xiu, Z.; Jin, N.; Lu, H.; Li, X.; et al. Myricetin activates the caspase-3/GSDME pathway via ER stress induction of pyroptosis in lung cancer cells. Front. Pharmacol. 2022, 13, 959938. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, S.; Oertlin, C.; Szczepanowska, K.; Kukat, A.; Senft, K.; Lucas, C.; Brodesser, S.; Hatzoglou, M.; Larsson, O.; Topisirovic, I.; et al. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 2021, 7, eabf0971. [Google Scholar] [CrossRef]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of cell death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Vejux, A.; Abed-Vieillard, D.; Hajji, K.; Zarrouk, A.; Mackrill, J.J.; Ghosh, S.; Nury, T.; Yammine, A.; Zaibi, M.; Mihoubi, W.; et al. 7-Ketocholesterol and 7β-hydroxycholesterol: In vitro and animal models used to characterize their activities and to identify molecules preventing their toxicity. Biochem. Pharmacol. 2020, 173, 113648. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Villani, R.; Tamborra, R.; Blonda, M.; Iannelli, G.; di Bello, G.; Facciorusso, A.; Poli, G.; Iuliano, L.; Avolio, C.; et al. Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 2018, 15, 86–96. [Google Scholar] [CrossRef] [PubMed]
- de Dios, C.; Abadin, X.; Roca-Agujetas, V.; Jimenez-Martinez, M.; Morales, A.; Trullas, R.; Mari, M.; Colell, A. Inflammasome activation under high cholesterol load triggers a protective microglial phenotype while promoting neuronal pyroptosis. Transl. Neurodegener. 2023, 12, 10. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Zhang, J.; Cai, L.; Guo, L.; Cai, Z.; Han, H.; Zhang, W. Cholestane-3β,5α,6β-triol Induces Multiple Cell Death in A549 Cells via ER Stress and Autophagy Activation. Mar. Drugs 2024, 22, 174. https://doi.org/10.3390/md22040174
Chen J, Zhang J, Cai L, Guo L, Cai Z, Han H, Zhang W. Cholestane-3β,5α,6β-triol Induces Multiple Cell Death in A549 Cells via ER Stress and Autophagy Activation. Marine Drugs. 2024; 22(4):174. https://doi.org/10.3390/md22040174
Chicago/Turabian StyleChen, Jiaxi, Jieping Zhang, Lijuan Cai, Li Guo, Zhenyu Cai, Hua Han, and Wen Zhang. 2024. "Cholestane-3β,5α,6β-triol Induces Multiple Cell Death in A549 Cells via ER Stress and Autophagy Activation" Marine Drugs 22, no. 4: 174. https://doi.org/10.3390/md22040174
APA StyleChen, J., Zhang, J., Cai, L., Guo, L., Cai, Z., Han, H., & Zhang, W. (2024). Cholestane-3β,5α,6β-triol Induces Multiple Cell Death in A549 Cells via ER Stress and Autophagy Activation. Marine Drugs, 22(4), 174. https://doi.org/10.3390/md22040174