Cyclodepsipeptides from Marine Sponges: Natural Agents for Drug Research

Abstract
:1. Introduction
2. Compounds with Anti-HIV Activity
2.1. Structural characteristics
2.2. Mode of action
2.3. Structure-activity relationship
3. Compounds with Anti-tumor Activity
3.1. Structural features
3.2. Mode of action
3.2.1. Actin polymerization
3.2.2. Polyploidization
3.2.3. Apoptosis
3.3. Jasplakinolide and arenastatin A analogs as mechanistic probes
4. Cyclodepsipeptides as Pharmacological Tools
4.1. Tools for studying actin organization and dynamics
4.2. Tools for studying the role of inhibition of Na+/Ca2+ exchange
4.3. Tools for studying cAMP-dependent transepithelial Cl− secretion
5. Conclusions
References
- Jimeno, JM. A clinical armamentarium of marine-derived anti-cancer compounds. Anticancer Drugs 2002, 13(Suppl 1), S15–19. [Google Scholar]
- Vera, MD; Joullié, MM. Natural products as probes of cell biology. Med Res Rev 2002, 22(2), 102–145. [Google Scholar]
- Davies, JS. The cyclization of peptides and depsipeptides. J Pept Sci 2003, 9(8), 471–501. [Google Scholar]
- Sarabia, F; Chammaa, S; Ruiz, AS; Ortiz, LM; Herrera, FJ. Chemistry and biology of cyclic depsipeptides of medicinal and biological interest. Curr Med Chem 2004, 11(10), 1309–1332. [Google Scholar]
- Hamada, Y; Shioiri, T. Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides. Chem Rev 2005, 105(12), 4441–4482. [Google Scholar]
- Sladic, D; Gasic, MJ. Reactivity and biological activity of the marine sesquiterpene hydroquinone avarol and related compounds from sponges of the order Dictyoceratida. Molecules 2006, 11(1), 1–33. [Google Scholar]
- Gupta, L; Talwar, A; Chauhan, PM. Bis and tris indole alkaloids from marine organisms, new leads for drug discovery. Curr Med Chem 2007, 14(16), 1789–1803. [Google Scholar]
- Liu, Y; Wang, L; Jung, JH; Zhang, S. Sesterterpenoids. Nat Prod Rep 2007, 24(6), 1401–1429. [Google Scholar]
- Fotie, J; Morgan, RE. Depsipeptides from microorganisms: a new class of antimalarials. Mini Rev Med Chem 2008, 8(11), 1088–1094. [Google Scholar]
- Araki, M; Miyake, S; Yamamura, T. Synthetic glycolipid ligands for human iNKT cells as potential therapeutic agents for immunotherapy. Curr Med Chem 2008, 15(23), 2337–2345. [Google Scholar]
- Godo, M; Sessler, T; Hamar, P. Role of invariant natural killer T (iNKT) cells in systemic lupus erythematosus. Curr Med Chem 2008, 15(18), 1778–1787. [Google Scholar]
- Skropeta, D. Deep-sea natural products. Nat Prod Rep 2008, 25(6), 1131–1166. [Google Scholar]
- Taylor, RE. Tedanolide and the evolution of polyketide inhibitors of eukaryotic protein synthesis. Nat Prod Rep 2008, 25(5), 854–861. [Google Scholar]
- Blunt, JW; Copp, BR; Hu, WP; Munro, MHG; Northcote, PT; Prinsep, MR. Marine natural products. Nat Prod Rep 2008, 25(1), 35–94. [Google Scholar]
- Provencio, M; Sánchez, A; Gasent, J; Gómez, P; Rosell, R. Cancer treatments: Can we find treasures at the bottom of the sea. Clin Lung Cancer 2009, 10(4), 295–300. [Google Scholar]
- Barnathan, G. Non-methylene-interrupted fatty acids from marine invertebrates: Occurrence, characterization and biological properties. Biochimie 2009, 91(6), 671–678. [Google Scholar]
- Fusetani, N; Kem, W. Marine toxins: an overview. Prog Mol Subcell Biol 2009, 46, 1–44. [Google Scholar]
- Gademann, K; Kobylinska, J. Antimalarial natural products of marine and freshwater origin. Chem Rec 2009, 9(3), 187–198. [Google Scholar]
- Laport, MS; Santos, OC; Muricy, G. Marine sponges: potential sources of new antimicrobial drugs. Curr Pharm Biotechnol 2009, 10(1), 86–105. [Google Scholar]
- Paterson, I; Dalby, SM. Synthesis and stereochemical determination of the spirastrellolides. Nat Prod Rep 2009, 26(7), 865–873. [Google Scholar]
- Wu, L; Gabriel, CL; Parekh, VV; Van Kaer, L. Invariant natural killer T cells: innate-like T cells with potent immunomodulatory activities. Tissue Antigens 2009, 73(6), 535–545. [Google Scholar]
- Munoz-Alonso, MJ; González-Santiago, L; Martínez, T; Losada, A; Galmarini, CM; Munoz, A. The mechanism of action of plitidepsin. Curr Opin Investig Drugs 2009, 10(6), 536–542. [Google Scholar]
- Andjelic, CD; Planelles, V; Barrows, LR. Characterizing the anti-HIV activity of papuamide A. Mar Drugs 2008, 6(4), 528–549. [Google Scholar]
- Plaza, A; Gustchina, E; Baker, HL; Kelly, M; Bewley, CA. Mirabamides A-D, depsipeptides from the sponge Siliquariaspongia mirabilis that inhibit HIV-1 fusion. J Nat Prod 2007, 70(11), 1753–1760. [Google Scholar]
- Tan, JJ; Cong, XJ; Hu, LM; Wang, CX; Jia, L; Liang, XJ. Therapeutic strategies underpinning the development of novel techniques for the treatment of HIV infection. Drug Discov Today 2010, 15, 186–197. [Google Scholar]
- Hamann, MT; Otto, CS; Scheuer, PJ; Dunbar, DC. Kahalalides: Bioactive peptides from a marine mollusk Elysia rufescens and its algal diet bryopsis sp. J Org Chem 1996, 61, 6594–6600. [Google Scholar]
- Rademaker-Lakhai, JM; Horenblas, S; Meinhardt, W; Stokvis, E; de Reijke, TM; Jimeno, JM; Lopez-Lazaro, L; Lopez Martin, JA; Beijnen, JH; Schellens, JH. Phase I clinical and pharmacokinetic study of kahalalide F in patients with advanced androgen refractory prostate cancer. Clin Cancer Res 2005, 11, 1854–1862. [Google Scholar]
- Pardo, B; Paz-Ares, L; Tabernero, J; Ciruelos, E; Garcia, M; Salazar, R; López, A; Blanco, M; Jimeno, J; Izquierdo, MA; Triqo, JM. Phase I clinical and pharmacokinetic study of kahalalide F administered weekly as a 1-hour infusion to patients with advanced solid tumors. Clin Cancer Res 2008, 14, 1116–1123. [Google Scholar]
- Martin-Algara, S; Espinosa, E; Rubió, J; López, JJ; Manzano, JL; Plazaola, A; Tanovic, A; Paz-Ares, L. Phase II study of weekly kahalalide F in patients with advanced malignant melanoma. Eur J Cancer 2009, 45(5), 732–735. [Google Scholar]
- Faivre, S; Chièze, S; Delbaldo, C; Ady-vago, N; Guzman, C; Lopez-Lazaro, L; Lozahic, S; Jimeno, J; Pico, F; Armand, JP; Lopez Martin, JA; Raymond, E. Phase I and Pharmacokinetic Study of Aplindine, a New Marine Cyclodepsipeptide in Patients With Advanced Malignancies. J Clin Oncol 2005, 23, 7871–7880. [Google Scholar]
- Maroun, JA; Belanger, K; Seymour, L; Matthews, S; Roach, J; Dionne, J; Soulieres, D; Stewart, D; Goel, R; Charpentier, D; Goss, G; Tomiak, E; Yau, J; Jimeno, J; Chiritescu, G. Phase I study of Aplidine in a dailyx5 one-hour infusion every 3 weeks in patients with solid tumors refractory to standard therapy. A National Cancer Institute of Canada Clinical Trials Group study, NCIC CTG IND 115. Ann Oncol 2006, 17, 1371–1378. [Google Scholar]
- Mitsiades, CS; Ocio, EM; Pandiella, A; Maiso, P; Gajate, C; Garayoa, M; Vilanova, D; Montero, JC; Mitsiades, N; McMullan, CJ; Munshi, NC; Hideshima, T; Chauhan, D; Aviles, P; Otero, G; Faircloth, G; Mateos, MV; Richardson, PG; Mollinedo, F; San-Miguel, JF; Anderson, KC. Aplidin, a marine organism-derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res 2008, 68(13), 5216–5225. [Google Scholar]
- Peschel, C; Hartmann, JT; Schmittel, A; Bokemeyer, C; Schneller, F; Keilholz, U; Buchheidt, D; Millan, S; Izquierdo, MA; Hofheinz, RD. Phase II study of plitidepsin in pretreated patients with locally advanced or metastatic non-small cell lung cancer. Lung Cancer 2008, 60, 374–380. [Google Scholar]
- Izqierdo, MA; Bowman, A; García, M; Jodrell, D; Martinez, M; Pardo, B; Gómez, J; López-Martin, JA; Jimeno, J; Germá, JR; Smyth, JF. Phase I clinical and pharmacokinetic study of plitidepsin as a 1-hour weekly intravenous infusion in patients with advanced solid tumors. Clin Cancer Res 2008, 14, 3105–3112. [Google Scholar]
- Nalda-Molina, R; Valenzuela, B; Ramon-Lopez, A; Miguel-Lillo, B; Soro-Matos, A; Perez-Ruixo, JJ. Population pharmacokinetics meta-analysis of plitidepsin (Aplidin®) in cancer subjects. Cancer Chemother Pharmacol 2009, 64(1), 97–108. [Google Scholar]
- Morton, CL; Houghton, PJ; Gorlick, R; Kolb, EA; Lock, R; Carol, H; Keir, ST; Reynolds, CP; Kang, MH; Maris, JM; Billups, C; Smith, MA. Initial testing of aplidin by the pediatric pre-clinical testing program. Pediatr Blood Cancer 2009, 53(3), 509–512. [Google Scholar]
- Dumez, H; Gallardo, E; Culine, S; Galceran, JC; Schöffski, P; Droz, JP; Extremera, S; Szyldergemajn, S; Fléchon, A. Phase II study of biweekly plitidepsin as second-line therapy for advanced or metastatic ttransitional cell carcinoma of the urothelium. Mar Drugs 2009, 7(3), 451–463. [Google Scholar]
- Rockwell, S; Liu, Y. Aplidin as a potential adjunct to radiation therapy: in vitro studies. Int J Radiat Biol 2010, 86(1), 63–70. [Google Scholar]
- Liang, B; Carroll, PJ; Joullie, MM. Progress toward the total synthesis of callipeltin A (I): asymmetric synthesis of (3S,4R)-3,4-dimethylglutamine. Org Lett 2000, 2(26), 4157–4160. [Google Scholar]
- Thoen, JC; Morales-Ramos, ÁI; Lipton, MA. Synthesis of the unnatural amino acid AGDHE, a constituent of the cyclic ddepsipeptides callipeltins A and D. Org Lett 2002, 4(25), 4455–4458. [Google Scholar]
- Turk, JA; Visbal, GS; Lipton, MA. Asymmetric synthesis of four diastereomers of 3-hydroxy-2,4,6-trimethylheptanoic acid: proof of configurational assignment. J Org Chem 2003, 68(20), 7841–7844. [Google Scholar]
- Okamoto, N; Hara, O; Makino, K; Hamada, Y. Diastereoselective synthesis of all stereoisomers of beta-methoxytyrosine, a component of papuamides. J Org Chem 2002, 67(26), 9210–9215. [Google Scholar]
- Hansen, DB; Wan, X; Carroll, PJ; Joullié, MM. Stereoselective synthesis of four stereoisomers of beta-methoxytyrosine, a component of callipeltin A. J Org Chem 2005, 70(8), 3120–3126. [Google Scholar]
- Oku, N; Gustafson, KR; Cartner, LK; Wilson, JA; Shigematsu, N; Hess, S; Pannell, LK; Boyd, MR; McMahon, JB. Neamphamide A, a new HIV-inhibitory depsipeptide from the Papua New Guinea marine sponge Neamphius huxleyi. J Nat Prod 2004, 67(8), 1407–1411. [Google Scholar]
- Zampella, A; D’Orsi, R; Sepe, V; Casapullo, A; Monti, MC; D’Auria, MV. Concise synthesis of all stereoisomers of beta-methoxytyrosine and determination of the absolute configuration of the residue in callipeltin A. Org Lett 2005, 7(16), 3585–3588. [Google Scholar]
- Calimsiz, S; Morales Ramos, AI; Lipton, MA. Solid-phase synthesis and configurational reassigment of callipeltin E. Implications for the structures of callipeltins A and B. J Org Chem 2006, 71(17), 6351–6356. [Google Scholar]
- Krishnamoorthy, R; Vasquez-Serrano, LD; Turk, JA; Kowalski, JA; Benson, AG; Breaux, NT; Lipton, MA. Solid-phase total synthesis and structure proof of callipeltin B. J Am Chem Soc 2006, 128(48), 15392–15393. [Google Scholar]
- Xie, W; Ding, D; Zi, W; Li, G; Ma, D. Total synthesis and structure assignment of papuamide B, a potent marine cyclodepsipeptide with anti-HIV properties. Angew Chem Int Ed Engl 2008, 47(15), 2844–2848. [Google Scholar]
- Zampella, A; D’Auria, V; Gomez Paloma, L; Casapullo, A; Minale, L; Debitus, C; Henin, Y. Callipeltin A, an anti-HIV cyclic depsipeptide from the new Caledonian Listhistida sponge Calipellta sp. J Am Chem Soc 1996, 118, 6202–6209. [Google Scholar]
- Plaza, A; Bifulco, G; Keffer, JL; Lloyd, JR; Baker, HL; Bewley, CA. Celebesides A–C and theopapuamides B–D, depsipeptides from an Indonesian sponge that inhibit HIV-1 entry. J Org Chem 2009, 74(2), 504–512. [Google Scholar]
- Zampella, A; Sepe, V; Luciano, P; Bellotta, F; Monti, MC; D’Auria, MV; Jepsen, T; Petek, S; Adeline, MT; Laprévôte, O; Aubertin, AM; Debitus, C; Poupat, C; Ahond, A. Homophymine A, an anti-HIV cyclodepsipeptide from the sponge Homophymia sp. J Org Chem 2008, 73(14), 5319–5327. [Google Scholar]
- Rashid, MA; Gustafson, KR; Cartner, LK; Shigematsu, N; Pannell, LK; Boyd, MR. Microspinosamide, a new HIV-inhibitory cyclic depsipeptide from the marine sponge Sidonops microspinosa. J Nat Prod 2001, 64(1), 117–121. [Google Scholar]
- Ford, PW; Gustafson, KR; McKee, TC; Shigematsu, N; Maurizi, LK; Pannell, LK; Williams, DE; Dilip de Silva, E; Lassota, P; Allen, TM; Van Soest, R; Andersen, RJ; Boyd, MR. Papuamides A–D, HIV inhibitory and cytotoxic depsipeptides from the sponges Theonella mirabilis and Theonella swinhoei collected in Papua New Guinea. J Am Chem Soc 1999, 121, 5899–5909. [Google Scholar]
- Oku, N; Krishnamoorthy, R; Benson, AG; Ferguson, RL; Lipton, MA; Phillips, LR; Gustafson, KR; McMahon, JB. Complete stereochemistry of neamphamide A and absolute configuration of the beta-methoxytyrosine residue in papuamide B. J Org Chem 2005, 70(17), 6842–6847. [Google Scholar]
- Ratnayake, AS; Bugni, TS; Feng, X; Harper, MK; Skalicky, JJ; Mohammed, KA; Andjelic, CD; Barrows, LR; Ireland, CM. Theopapuamide, a cyclic depsipeptide from a Papua New Guinea lithistid sponge Theonella swinhoei. J Nat Prod 2006, 69(11), 1582–1586. [Google Scholar]
- Dalgleish, AG. The pathogenesis of AIDS: classical and alternative views. J R Coll Physicians Lond 1992, 26(2), 152–158. [Google Scholar]
- McDougal, JS; Klatzmann, DR; Maddon, PJ. CD4-gp120 interactions. Curr Opin Immunol 1991, 4, 552–558. [Google Scholar]
- Rizzuto, CD; Wyatt, R; Hernández-Ramos, N; Sun, Y; Kwong, PD; Hendrickson, WA; Sodroski, J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 1998, 280(5371), 1949–1953. [Google Scholar]
- Liu, S; Wu, S; Jiang, S. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr Pharm Des 2007, 13(2), 143–162. [Google Scholar]
- Doms, RW; Moore, JP. HIV-1 membrane fusion: targets of opportunity. J Cell Biol 2000, 151(2), F9–14. [Google Scholar]
- Chan, DC; Fass, D; Berger, JM; Kim, PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89(2), 263–273. [Google Scholar]
- Tan, K; Liu, J; Wang, J; Shen, S; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci USA 1997, 94(23), 12303–12308. [Google Scholar]
- Weissenhorn, W; Dessen, A; Harrison, SC; Skehel, JJ; Wiley, DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387(6631), 426–430. [Google Scholar]
- Volpon, L; Besson, F; Lancelin, JM. NMR structure of active and inactive forms of the sterol-dependent antifungal antibiotic bacillomycin L. Eur J Biochem 1999, 264(1), 200–210. [Google Scholar]
- Campbell, SM; Crowe, SM; Mak, J. Virion-associated cholesterol is critical for the maintenance of HIV-1 structure and infectivity. AIDS 2002, 16(17), 2253–2261. [Google Scholar]
- Raulin, J. Human immunodeficiency virus and host cell lipids. Interesting pathways in research for a new HIV therapy. Prog Lipid Res 2002, 41(1), 27–65. [Google Scholar]
- Hayot, C; Debeir, O; Van Ham, P; Van Damme, M; Kiss, R; Decaestecker, C. Characterization of the activities of actin-affecting drugs on tumor cell migration. Toxicol Appl Pharmacol 2006, 211(1), 30–40. [Google Scholar]
- Terracciano, S; Bruno, I; D’Amico, E; Bifulco, G; Zampella, A; Sepe, V; Smith, CD; Riccio, R. Synthetic and pharmacological studies on new simplified analogues of the potent actin-targeting Jaspamide. Bioorg Med Chem 2008, 16(13), 6580–6588. [Google Scholar]
- Tabudravu, JN; Morris, LA; Milne, BF; Jaspars, M. Conformational studies of free and Li+ complexed jasplakinolide, a cyclic depsipeptide from the Fijian marine sponge Jaspis splendens. Org Biomol Chem 2005, 3(5), 745–749. [Google Scholar]
- Trevisi, L; Bova, S; Cargnelli, G; Danieli-Betto, D; Floreani, M; Germinario, E; D’Auria, MV; Luciani, S. Callipeltin A, a cyclic depsipeptide inhibitor of the cardiac sodium-calcium exchanger and positive inotropic agent. Biochem Biophys Res Commun 2000, 279(1), 219–222. [Google Scholar]
- Krishnamoorthy, R; Richardson, BL; Lipton, MA. Synthesis and cytotoxicity of desmethoxycallipeltin B: lack of a quinone methide for the cytotoxicity of callipeltin B. Bioorg Med Chem Lett 2007, 17(18), 5136–5138. [Google Scholar]
- Grassia, A; Bruno, I; Debitus, C; Marzocco, S; Pinto, A; Gomez-Paloma, L; Riccio, R. Spongidepsin, a new cytotoxic macrolide from Spongia sp. Tetrahedron 2001, 57, 6257–6260. [Google Scholar]
- Ferrie, L; Reymond, S; Capdevielle, P; Cossy, J. Total synthesis of (−)-spongidepsin. Org Lett 2006, 8(16), 3441–3443. [Google Scholar]
- Ghosh, A; Xu, X. Assignment of absolute stereochemistry and total synthesis of (−)-spongidepsin. Org Lett 2004, 6(12), 2055–2058. [Google Scholar]
- Zhu, G; Negishi, E. Fully reagent-controlled asymmetric synthesis of (−)-spongidepsin via the Zr-catalyzed asymmetric carboalumination of alkenes (ZACA reaction). Org Lett 2007, 9(15), 2771–2774. [Google Scholar]
- Zampella, A; Sepe, V; Bellotta, F; Luciano, P; D’Auria, MV; Cresteil, T; Debitus, C; Petek, S; Poupat, C; Ahond, A. Homophymines B–E and A1–1 a family of bioactive cyclodepsipeptides from the sponge Homophymia sp. Org Biomol Chem 2009, 7(19), 4037–4044. [Google Scholar]
- Stehn, JR; Schevzoy, G; O’Neill, GM; Gunning, PW. Specialisation of the tropomyosin composition of actin filaments provides new potential targets for chemotherapy. Curr Cancer Drug Targets 2006, 6(3), 245–256. [Google Scholar]
- Merajver, SD; Usmani, SZ. Multifaceted role of Rho proteins in angiogenesis. J Mammary Gland Biol Neoplasia 2005, 10(4), 291–198. [Google Scholar]
- Dummler, B; Ohshiro, K; Kumar, R; Field, J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev 2009, 28 . [Google Scholar]
- Bubb, MR; Senderowicz, AMJ; Sausville, EA; Duncan, KLK; Korn, ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem 1994, 269(21), 14869–14871. [Google Scholar]
- Ridley, AJ. Rho-related proteins: actin cytoskeleton and cell cycle. Curr Opin Genet Dev 1995, 5(1), 24–30. [Google Scholar]
- Fabian, I; Shur, I; Bleiberg, I; Rudi, A; Kashman, Y; Lishner, M. Growth modulation and differentiation of acute myeloid leukemia cells by jaspamide. Exp Hematol 1995, 23(7), 583–587. [Google Scholar]
- Fabian, I; Halperin, D; Lefter, S; Mittelman, L; Altstock, RT; Seaon, O; Tsarfaty, I. Alteration of actin organization by jaspamide inhibits ruffling, but not phagocytosis or oxidative burst, in HL-60 cells and human monocytes. Blood 1999, 93(11), 3994–4005. [Google Scholar]
- Bubb, MR; Spector, I; Beyer, BB; Fosen, KM. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J Biol Chem 2000, 275(7), 5163–5170. [Google Scholar]
- Ou, GS; Chen, ZL; Yuan, M. Jasplakinolide reversibly disrupts actin filaments in suspension-cultured tobacco BY-2 cells. Protoplasma 2002, 219 . [Google Scholar]
- Tinto, WF; Lough, AJ; McLean, S; Reynolds, WF; Yu, M; Chan, WR. Geodiamolides H and I, further cyclodepsipeptides from the marine sponge Geodia sp. Tetrahedron 1998, 54(18), 4451–4458. [Google Scholar]
- Rangel, M; Prado, M; Konno, K; Naoki, H; Freitas, JC; Machado-Santelli, GM. Cytoskeleton alterations induced by Geodia corticostylifera depsipeptides in breast cancer cells. Peptides 2006, 27(9), 2047–2057. [Google Scholar]
- Freitas, VM; Rangel, M; Bisson, LF; Jaeger, RG; Machado-Santelli, GM. The geodiamolide H, derived from Brazilian sponge Geodia corticostylifera, regulates actin cytoskeleton, migration and invasion of breast cancer cells cultured in three-dimensional environment. J Cell Physiol 2008, 216(3), 583–594. [Google Scholar]
- Nakazawa, H; Kitano, K; Cioca, D; Ishikawa, M; Ueno, M; Ishida, F; Kiyosawa, K. Induction of polyploidization by jaspamide in HL-60 cells. Acta Haematol 2000, 104 . [Google Scholar]
- Odaka, C; Sanders, ML; Crews, P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin Diagn Lab Immunol 2000, 7(6), 947–952. [Google Scholar]
- Cioca, DP; Kitano, K. Induction of apoptosis and CD10/neutral endopeptidase expression by jaspamide in HL-60 line cells. Cell Mol Life Sci 2002, 59(8), 1377–1387. [Google Scholar]
- Kobayashi, M; Wang, W; Ohyabu, N; Kurosu, M; Kitagawa, I. Improved total synthesis and structure-activity relationship of arenastatin A, a potent cytotoxic spongean depsipeptide. Chem Pharm Bull (Tokyo) 1995, 43(9), 1598–1600. [Google Scholar]
- Koiso, Y; Morita, K; Kobayashi, M; Wang, W; Ohyabu, N; Iwasaki, S. Effects of arenastatin A and its synthetic analogs on microtubule assembly. Chem Biol Interact 1996, 102(3), 183–191. [Google Scholar]
- Morita, K; Koiso, Y; Hashimoto, Y; Kobayashi, M; Wang, W; Ohyabu, N; Iwasaki, S. Interaction of arenastatin A with porcine brain tubulin. Biol Pharm Bull 1997, 20(2), 171–174. [Google Scholar]
- Kotoku, N; Kato, T; Narumi, F; Ohtani, E; Kamada, S; Aoki, S; Okada, N; Nakagawa, S; Kobayashi, M. Synthesis of 15,20-triamide analogue with polar substituent on the phenyl ring of arenastatin A, an extremely potent cytotoxic spongean depsipeptide. Bioorg Med Chem 2006, 14(22), 7446–7457. [Google Scholar]
- Rao, JY; Jin, YS; Zheng, QL; Cheng, J; Tai, J; Hemstreet, GP, III. Alterations of the actin polymerization status as an apoptotic morphological effector in HL-60 cells. J Cell Biochem 1999, 75(4), 686–697. [Google Scholar]
- Marimganti, S; Yasmeen, S; Fischer, D; Maier, M. Synthesis of jasplakinolide analogues containing a novel omega-amino acid. Chemistry 2005, 11(22), 6687–6700. [Google Scholar]
- Hu, TS; Tannert, R; Arndt, HD; Waldmann, H. Solid-phase based synthesis of jasplakinolide analogs by intramolecular azide-alkyne cycloadditions. Chem Commun (Camb) 2007, 3942–3944. [Google Scholar]
- Terracciano, S; Bruno, I; Bifulco, G; Avallone, E; Smith, CD; Gomez-Paloma, L; Riccio, R. Synthesis, solution structure, and bioactivity of six new simplified analogues of the natural cyclodepsipeptide jaspamide. Bioorg Med Chem 2005, 13(17), 5225–5239. [Google Scholar]
- Eggen, M; Mossman, CJ; Buck, SB; Nair, SK; Bhat, L; Ali, SM; reiff, EA; Boge, TC; Georg, GI. Total synthesis of cryptophycin-24 (Arenastatin A) amenable to structural modifications in the C16 side chain. J Org Chem 2000, 65(23), 7792–7799. [Google Scholar]
- Murakami, N; Wang, W; Tamura, S; Kobayashi, M. Synthesis and biological property of carba and 20-deoxo analogues of arenastatin A. Bioorg Med Chem Lett 2000, 10(16), 1823–1826. [Google Scholar]
- Murakami, N; Tamura, S; Koyama, K; Sugimoto, M; Maekawa, R; Kobayashi, M. New analogue of arenastatin A, a potent cytotoxic spongean depsipeptide, with anti-tumor activity. Bioorg Med Chem Lett 2004, 14(10), 2597–2601. [Google Scholar]
- Shaw, MK; Tilney, LG. Induction of an acrosomal process in Toxoplasma gondii: visualization of actin filaments in a protozoan parasite. Proc Natl Acad Sci USA 1999, 96(16), 9095–9009. [Google Scholar]
- Terada, Y; Simerly, C; Schatten, G. Microfilament stabilization by jasplakinolide arrests oocyte maturation, cortical granule exocytosis, sperm incorporation cone resorption, and cell-cycle progression, but not DNA replication, during fertilization in mice. Mol Reprod Dev 2000, 56(1), 89–98. [Google Scholar]
- Di Campli, A; Valderrama, F; Babià, T; De Matteis, MA; Liuni, A; Egea, G. Morphological changes in the Golgi complex correlate with actin cytoskeleton rearrangements. Cell Motil Cytoskeleton 1999, 43(4), 334–348. [Google Scholar]
- Cramer, LP. Role of actin-filament disassembly in lamellipodium protrusion in motile cells revealed using the drug jasplakinolide. Curr Biol 1999, 9(19), 1095–1105. [Google Scholar]
- Posey, SC; Bierer, BE. Actin stabilization by jasplakinolide enhances apoptosis induced by cytokine deprivation. J Biol Chem 1999, 274(7), 4259–4265. [Google Scholar]
- Holzinger, A; Meindl, U. Jasplakinolide, a novel actin targeting peptide, inhibits cell growth and induces actin filament polymerization in the green alga Micrasterias. Cell Motil Cytoskeleton 1997, 38(4), 365–372. [Google Scholar]
- Trevisi, L; Cargnelli, G; Ceolotto, G; Papparella, I; Semplicini, A; Zampella, A; D’Auria, MV; Luciani, S. Callipeltin A: sodium ionophore effect and tension development in vascular smooth muscle. Biochem Pharmacol 2004, 68(7), 1331–1338. [Google Scholar]
- Matthews, JB; Smith, JA; Hrnjez, BJ. Effects of F-actin stabilization or disassembly on epithelial Cl- secretion and Na-K-2Cl cotransport. Am J Physiol 1997, 272, C254–262. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Cyclo-Depsipeptide | Marine Sponge | Uncommon Residues | ||
|---|---|---|---|---|
| Amino acids | Polyketide moieties | Ref. | ||
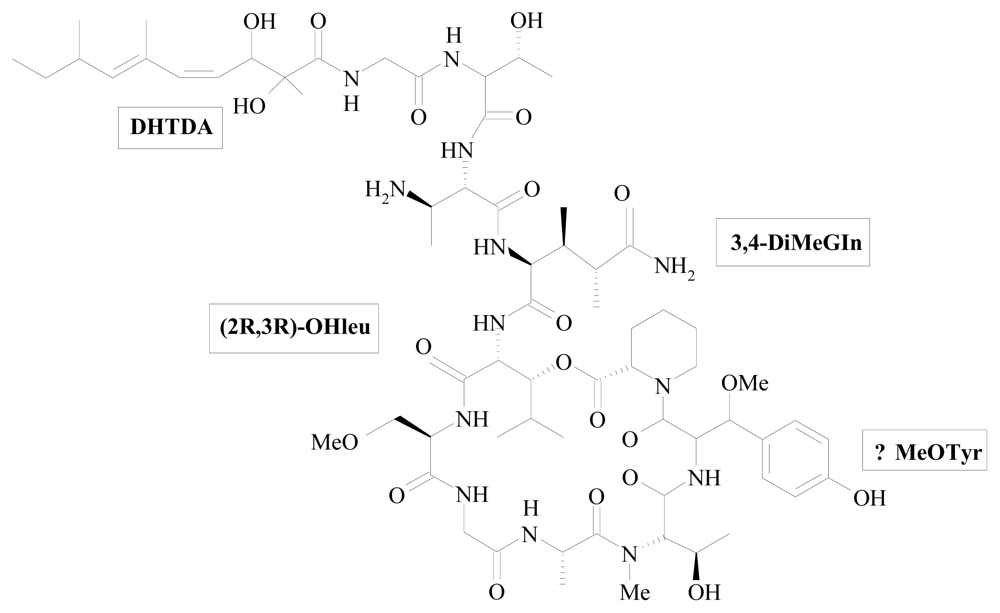
| Callipeltin A | Callipelta sp. | AGDHA 3,4-DiMeGln β-OMeTyr | [40] [43] [45] [49] | |
| Celebesides | Siliquariaspongia mirabilis | ACPA 3,4-DiMeGln 3-CThr pSer (Cel. A, B) | DDTD HTMOA | [50] |
| Homophymine A | Homophymia sp | ADHA AHDMHA 3,4-DiMeGln ThrOMe | HTMOA | [51] |
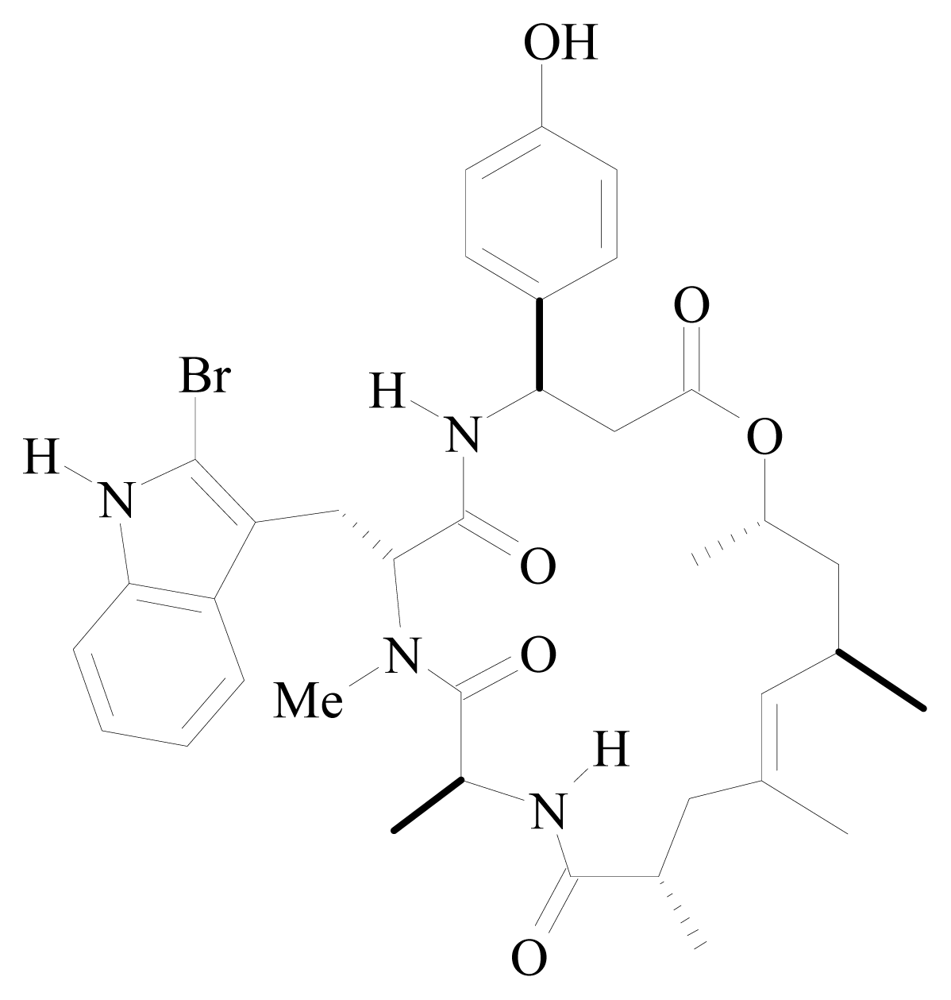
| Microspinosamide | Sidonops microspinosa | N-MeGln HBPA | [52] | |
| Mirabamides | Siliquariaspongia mirabilis | Dab (except M. B) 3,4-DiMeGln β-OMeTyrRP ClHPr | DHTDA | [24] |
| Neamphamide A | Neamphius huxleyi | AGDHA 3,4-DiMeGln N-MeGln β-OMeTyr | HTMHA | [44] |
| Papuamides | Theonella mirabilis and Theonella swinhoei | Dab 3,4-DiMeGln hPro β-OMeTyr 3-OHLeu | DHTDA | [48] [53] [54] |
| Theopapuamides | Geodia barretti and Siliquariaspongia mirabilis | ACPA AMDHA (Th. B) AMTHA (Th. A) 3,4-DiMeGln N-MeGln hiLeu (Th. C) | HTMOA | [55] |
| ACPA | 3-acetamido-2-aminopropanoic acid |
| ADHA | 4-amino-2,3-dihydroxy-1,7-heptanoic acid |
| AGDHA | 4-amino-7-guanidino-2,3-dihydroxyheptanoic acid |
| AHDMHA | 2-amino-3-hydroxy-4,5-dimethylhexanoic acid |
| AMDHA | 4-amino-2,3-dihydroxy-5-methylhexanoic acid |
| AMTHA | 4-amino-2,3,5-trihydroxy-5-methylhexanoic acid |
| ClHPr | 4-chlorohomoproline |
| 3-CThr | 3-carbamoyl threonine |
| Dab | 2,3-diaminobutanoic acid |
| DDTD | 7,9-dihydroxy-8,10-dimethyltrideca-2,4-dienoic acid |
| DHTDA | 2,3-dihydroxy-2,6,8-trimethyldeca-4,6-dienoic acid |
| 3,4-DiMeGln | 3,4-dimethyl-L-glutamine |
| hiLeu | homoisoleucine |
| hPro | homoproline |
| HBPA | β-hydroxy-p-bromophenylalanine |
| HTMOA | 3-hydroxy-2,4,6-trimethyloctanoic acid |
| HTMHA | 3-hydroxy-2,4,6-trimethylheptanoic acid |
| N-MeGln | N-Methylglutamin |
| β-OMeTyr | β-methoxytyrosine |
| β-OMeTyrRP | β-methoxytyrosine 4′-O-α-L-rhamnopyranoside |
| 3-OHLeu | 3-hydroxyleucine |
| pSer | phosphoserine |
| ThrOMe | O-methyltreonine |
| Cyclo-Depsipeptide | Assay | Anti-HIV Activity (IC50) | Cytotoxicity (TC50) | Ref. |
|---|---|---|---|---|
| Callipeltin A | MTT cell viability on CEM4 lymphocytic cell lines infected with HIV-1 (Lai strain, X4 tropic) | 0.01 μg/mL | 0.29 μg/mL | [49] |
| Celebesides | single-round HIV-1 infectivity assay against viruses pseudo-typed with HIV-1 SF162 Envelope | A: 1.9 μg/mL B: >50 μg/mL | [50] | |
| Homophymine A | production of HIV-1 virus (III B strain) measured by quantification of reverse transcriptase activity associated with the virus particles in PBMC cell lines | 75 nM | 1.19 μM | [51] |
| Microspinosamide | XTT-based cell viability assay in HIV-1 infected CEM-SS target cells | 0.2 μg/mL | 3.0 μg/mL | [52] |
| Mirabamides A-D | HIV-1 neutralization assays: | HXB2: | [24] | |
| HXB2 (T-cell-tropic) and SF162 (macrophage-tropic) viral strains; | A: 140 nM | |||
| TZM-bl host cells (expressing CXCR4, CCR5, and CD4) | B: >50 μM | |||
| C: 140 nM | ||||
| D: 190 nM | ||||
| SF162: | ||||
| A: 400 nM | ||||
| C, D: 1 μM | ||||
| HIV-1 envelope-mediated cell fusion assay | A: 41 nM | |||
| C, D: low μM range | ||||
| Neamphamide A | XTT-based cell viability assay: human T-cell line CEM-SS infected with HIV-1RF | 28 nM | 260 nM | [44] |
| Papuamide A | HIV-1 envelope-mediated cell fusion assay | 73 nM | [24] | |
| MTT cell viability test | 71 nM | [23] | ||
| Virion based fusion assay: co-transfection of pMM310 (plasmid encoding β-lactamase), pAdV Antage (vector), pNL4-3 (X4 tropic proviral DNA) or pNL(AD8) (R5 tropic proviral DNA) into 293FT cells; HeLaT4 or TZM-bl host cells | 114 nM | [23] | ||
| Pseudo-type virus assay: pDHIV-3 plus envelope glycoprotein plasmids transfected to 293FT cells; Target cells: CEM-SS or CEM.NKR-CCR5 cells | 178 nM | [23] | ||
| tetrazolium-based assay CEM-SS T-cells | 3.6 ng/mL | 74 ng/mL | [53] | |
| Theopapuamide B | single-round HIV-1 infectivity assay against viruses pseudo-typed with HIV-1 SF162 Envelope | 0.8 μg/mL | [50] | |
| IC50 | half-maximal concentration for cytoprotective activity against HIV-infection |
| TC50 | concentration for 50% reduction in cell viability (half-maximal concentration for cytotoxic response) |
| Cyclodepsipeptide | Marine sponge | Cell line | Growth inhibitory concentration (IC50) | Ref. |
|---|---|---|---|---|
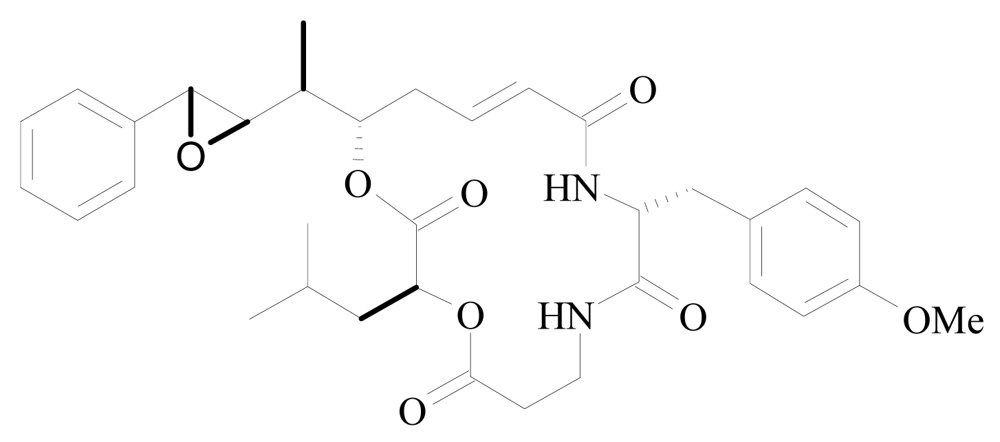
| Arenastatin A (cryptophycin –24) | Dysidea arenaria | KB 3-1 | 5 pg/mL | [92] [93] [94] |
| diethylamine analog | S180 | 0.18 ng/mL | [95] | |
| Geodiamolides | Geodia sp. and Geodia corticostylifera | [86] | ||
| sea urchin eggs; | ≈100–600 nM | [87] | ||
| T47D and MCF7 | ≈20–115 nM | [87] | ||
| Hs578T | 120 nM | [88] | ||
| Homophymines | Homophymia sp. | PC3, OV3, MCF7/MCF7R, HCT116/HCT15, HL60/HL60R | 2–100 nM | [76] |
| Jasplakinolide (jaspamide) | Jaspis sp. and Hemiastrella minor | HL-60 | 100 nM | [83] |
| 50 nM | [89] | |||
| 100 nM | [91] | |||
| 100 nM | [96] | |||
| Jurkat T cells, EL-4, SP-2/0, J774.1 | 2 μg/mL | [90] | ||
| Spongidepsin | Spongia sp. | J774.A1 | 0.56 μM | [72] |
| WEHI-164 | 0.42 μM | |||
| HEK-293 | 0.66 μM | |||
| Theopapuamides | Geodia barretti and Siliquariaspongia mirabilis | CEM-TART HCT-116 | 0.5 μM | [55] |
| 0.9 μM | ||||
| HCT-116 | Theop. A: 2.1 μg/mL | [50] | ||
| Theop. B: 4.0 μg/mL | ||||
| Theop. C: 2.1 μg/mL | ||||
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Andavan, G.S.B.; Lemmens-Gruber, R. Cyclodepsipeptides from Marine Sponges: Natural Agents for Drug Research. Mar. Drugs 2010, 8, 810-834. https://doi.org/10.3390/md8030810
Andavan GSB, Lemmens-Gruber R. Cyclodepsipeptides from Marine Sponges: Natural Agents for Drug Research. Marine Drugs. 2010; 8(3):810-834. https://doi.org/10.3390/md8030810
Chicago/Turabian StyleAndavan, Gowri Shankar Bagavananthem, and Rosa Lemmens-Gruber. 2010. "Cyclodepsipeptides from Marine Sponges: Natural Agents for Drug Research" Marine Drugs 8, no. 3: 810-834. https://doi.org/10.3390/md8030810
APA StyleAndavan, G. S. B., & Lemmens-Gruber, R. (2010). Cyclodepsipeptides from Marine Sponges: Natural Agents for Drug Research. Marine Drugs, 8(3), 810-834. https://doi.org/10.3390/md8030810