Microtubule-Stabilizing Drugs from Marine Sponges: Focus on Peloruside A and Zampanolide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Drugs from marine sources
1.2. Cytoskeletal targets
1.3. Microtubule-targeting drugs
2. Discussion
2.1. Peloruside A and its cell-mates, mycalamide A and pateamine
2.2. Mycalamides
2.3. Pateamine
2.4. Peloruside A
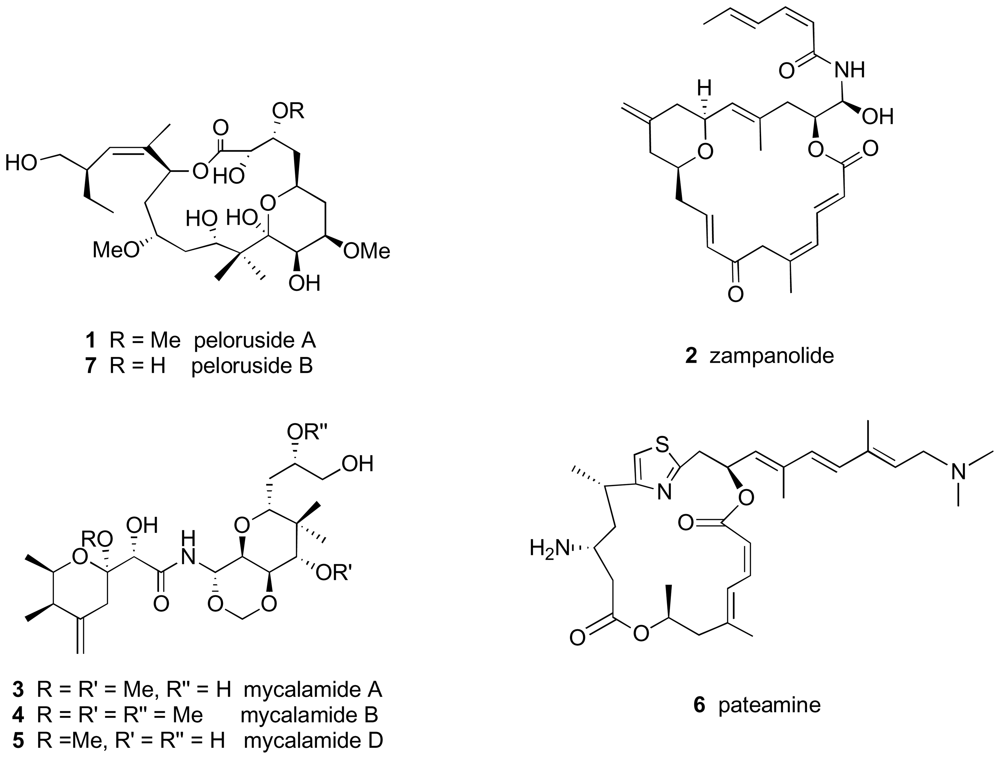
2.4.1. Discovery and mode of action
2.4.2. Peloruside A effects on cell proteins
2.4.3. Peloruside A binding site on tubulin
2.4.4. Peloruside congeners and analogues
2.4.5. Anti-disease potential of peloruside A
2.5. Zampanolide
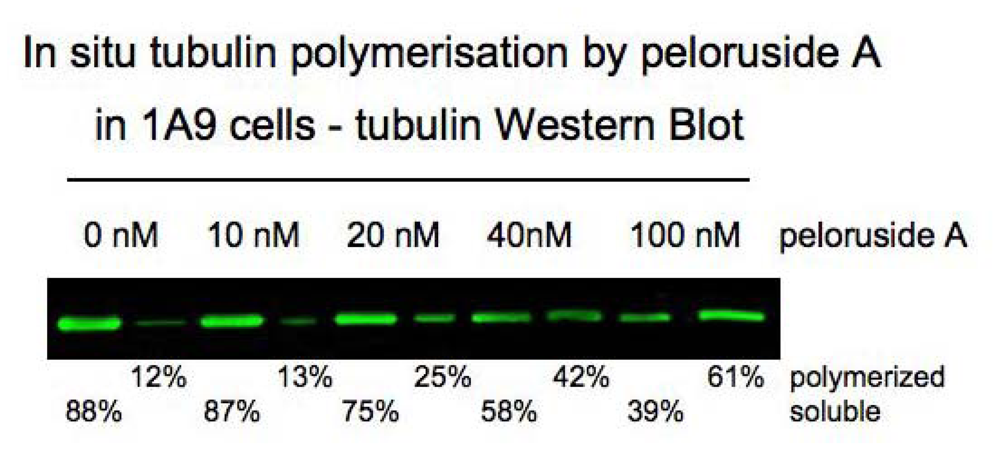
2.5.1. Discovery and synthesis
2.5.2. Mode of action
3. Conclusions
Acknowledgements
References
- Paterson, I; Anderson, EA. The renaissance of natural products as drug candidates. Science 2005, 310, 451–453. [Google Scholar]
- Newman, D; Cragg, GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod 2007, 70, 461–477. [Google Scholar]
- Molinski, TF; Dalisay, DS; Lievens, SL; Saludes, JP. Drug development from marine natural products. Nat Rev Drug Discov 2009, 8, 69–85. [Google Scholar]
- Cragg, GM; Grothaus, PG; Newman, DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev 2009, 109, 3012–3043. [Google Scholar]
- Sashidara, KV; White, KN; Crews, P. A selective account of the effective paradigms and significant outcomes in the discovery of inspirational marine natural products. J Nat Prod 2009, 72, 588–603. [Google Scholar]
- Napolitano, JG; Daranas, AH; Norte, M; Fernández, JJ. Marine macrolides, a promising source of antitumor compounds. Anti-Cancer Agents Med Chem 2009, 9, 122–137. [Google Scholar]
- Blunt, JW; Copp, BR; Hu, W-P; Munro, MHG; Northcote, PT; Prinsep, MR. Marine natural products. Nat Prod Rep 2009, 26, 170–244, and previous reviews in this series. [Google Scholar]
- Belarbi, EH; Contreras, Gomez A; Chisti, Y; García Camacho, F; Molina Grima, E. Producing drugs from marine sponges. Biotechnol Adv 2003, 21, 585–598. [Google Scholar]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov Today 2009, 8, 536–544. [Google Scholar]
- Mestel, R. Drugs from the sea. Discover 1999, 71–74. [Google Scholar]
- Simmons, TL; Andrianasolo, E; McPhail, K; Flatt, P; Gerwick, WH. Marine natural products as anticancer drugs. Mol Cancer Ther 2005, 4, 333–342. [Google Scholar]
- Singh, R; Sharma, M; Joshi, P; Rawat, DS. Clinical status of anti-cancer agents derived from marine sources. Anticancer Agents Med Chem 2008, 8, 603–617. [Google Scholar]
- Sipkema, D; Franssen, MCR; Osinga, R; Tramper, J; Wijffels, RH. Marine sponges as pharmacy. Mar Biotech 2005, 7, 142–162. [Google Scholar]
- Flam, F. Chemical prospectors scour the seas for promising drugs. Science 1994, 266, 1324–1325. [Google Scholar]
- Davidson, BS. New dimensions in natural products research: cultured marine microorgansims. Curr Opin Biotech 1995, 6, 284–291. [Google Scholar]
- Bentley, R. Microbial secondary metabolites play important roles in medicine: Prospects for discovery of new drugs. Perspect Biol Med 1997, 40, 364–395. [Google Scholar]
- Bewley, CA; Faulkner, DJ. Lithistid sponges: Star performers or hosts to the stars. Angew Chem Int Ed 1998, 37, 2162–2178. [Google Scholar]
- Fusetani, N; Kem, W. Marine toxins: an overview. Prog Mol Subcell Biol 2009, 46, 1–44. [Google Scholar]
- Page, M; West, L; Northcote, P; Battershill, C; Kelly, M. Spatial and temporal variability of cytotoxic metabolites in populations of the New Zealand sponge Mycale hentscheli. J Chem Ecol 2005, 31, 1161–1174. [Google Scholar]
- Page, MJ; Northcote, PT; Webb, VL; Mackey, S; Handley, SJ. Aquaculture trials for the production of biologically active metabolites in the New Zealand sponge Mycale hentscheli (Demospongiae: Poecilosclerida). Aquaculture 2005, 250, 256–269. [Google Scholar]
- Jordan, MA; Wilson, L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol 1998, 10, 123–130. [Google Scholar]
- Jordan, MA; Wilson, L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004, 4, 253–265. [Google Scholar]
- Zhou, J; Giannakakou, P. Targeting microtubules for cancer chemotherapy. Curr Med Chem–Anti-Cancer Agents 2005, 5, 65–71. [Google Scholar]
- Altmann, KH; Gertsch, J. Anticancer drugs from nature—natural products as a unique source of new microtubule-stabilizing agents. Nat Prod Rep 2007, 24, 327–357. [Google Scholar]
- Kingston, DGI. Tubulin-interactive natural products as anticancer agents. J Nat Prod 2009, 72, 507–515. [Google Scholar]
- Saito, S. Toxins affecting actin filaments and microtubules. Prog Mol Subcell Biol 2009, 46, 187–219. [Google Scholar]
- Rowinsky, EK; Donehower, RC. Paclitaxel (Taxol). New Engl J Med 1995, 332, 1004–1014. [Google Scholar]
- Rowinsky, EK; Calvo, E. Novel agents that target tublin and related elements. Semin Oncol 2006, 33, 421–435. [Google Scholar]
- Morris, PG; Fornier, MN. Microtubule active agents: Beyond the taxane frontier. Clin Cancer Res 2008, 14, 7167–7172. [Google Scholar]
- Wani, MC; Taylor, HL; Wall, ME; Coggon, P; McPhail, AT. Plant antitumor agents. VI. The isolation and structure of Taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc 1971, 93, 2325–2327. [Google Scholar]
- Schiff, PB; Fant, J; Horwitz, SB. Promotion of microtubule assembly in vitro by Taxol. Nature 1979, 277, 665–667. [Google Scholar]
- Tinley, TL; Randall-Hlubek, DA; Leal, RM; Jackson, EM; Cessac, JW; Quada, JC, Jr; Hemscheidt, TK; Mooberry, SL. Taccalonolides E and A: Plant-derived steroids with microtubule-stabilizing activity. Cancer Res 2003, 63, 3211–3220. [Google Scholar]
- Bollag, DM; McQueney, PA; Zhu, J; Hensens, O; Koupal, L; Leisch, J; Goetz, M; Lazarides, E; Woods, CM. Epothilones, a new class of microtubule-stabilizing agents with a Taxol-like mechanism of action. Cancer Res 1995, 55, 2325–2333. [Google Scholar]
- Lindel, T; Jensen, PR; Fenical, W; Long, BH; Casazza, AM; Carboni, J; Fairchild, CR. Eleutherobin, a new cytotoxin that mimics paclitaxel (Taxol) by stabilizing microtubules. J Am Chem Soc 1997, 119, 8744–8745. [Google Scholar]
- Long, BH; Carboni, JM; Wasserman, AJ; Cornell, LA; Casazza, AM; Jensen, PR; Lindel, T; Fenical, W; Fairchild, CR. Eleutherobin, a novel cytotoxic agent that induces tubulin polymerisation, is similar to paclitaxel (Taxol). Cancer Res 1998, 58, 1111–1115. [Google Scholar]
- D’Ambrosio, M; Guerriero, A; Pietra, F. Sarcodictyin A and sarcodictyin B, novel diterpenoidic alcohols esterified by (E)-N(1)-methylurocanic acid. Isolation from the Mediterranean stolonifer Sarcodictyon roseum. Helv Chim Acta 1987, 70, 2019–2027. [Google Scholar]
- Hamel, E; Sackett, DL; Vourloumis, D; Nicolaou, KC. The coral-derived natural products eleutherobin and sarcodictyins A and B: Effects on the assembly of purified tubulin with and without microtubule-associated proteins and binding at the polymer taxoid site. Biochemistry 1999, 38, 5490–5498. [Google Scholar]
- Hood, KA; West, LM; Northcote, PT; Berridge, MV; Miller, JH. Induction of apoptosis by the marine sponge (Mycale) metabolites, mycalamide A and pateamine. Apoptosis 2001, 6, 207–219. [Google Scholar]
- Hood, KA; Bäckström, BT; West, LM; Northcote, PT; Berridge, MV; Miller, JH. The novel cytotoxic sponge metabolite peloruside A, structurally similar to bryostatin-1, has unique bioactivity independent of protein kinase C. Anti-Cancer Drug Des 2001, 16, 155–166. [Google Scholar]
- Miller, JH; Rouwé, B; Gaitanos, TN; Hood, KA; Crume, KP; Bäckström, BT; La Flamme, AC; Berridge, MV; Northcote, PT. Peloruside A enhances apoptosis in H-ras-transformed cells and is cytotoxic to proliferating T cells. Apoptosis 2004, 9, 785–796. [Google Scholar]
- Perry, NB; Blunt, JW; Munro, MHG. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus. Mycale J Am Chem Soc 1988, 110, 4850–4851. [Google Scholar]
- Perry, NB; Blunt, JW; Munro, MHG; Thompson, AM. Antiviral and antitumor agents from a New Zealand sponge, Mycale sp. 2. Structures and solution conformations of mycalamides A and B. J Org Chem 1990, 55, 223–227. [Google Scholar]
- West, LM; Northcote, PT; Hood, KA; Miller, JH; Page, MJ. Mycalamide D, a new cytotoxic amide from the New Zealand marine sponge Mycale species. J Nat Prod 2000, 63, 707–709. [Google Scholar]
- Simpson, JS; Garson, MJ; Blunt, JW; Munro, MHG; Hooper, JNA. Mycalamides C and D, cytotoxic compounds from the marine sponge Stylinos n. species. J Nat Prod 2000, 63, 704–706. [Google Scholar]
- Cardani, C; Ghiringhelli, D; Mondelli, R; Quilico, A. The structure of pederin. Tetrahedron Lett 1965, 6, 2537–2545. [Google Scholar]
- Soldati, M; Fioretti, A; Ghione, M. Cytotoxicity of pederin and some of its derivatives on cultured mammalian cells. Experientia 1966, 22, 176–178. [Google Scholar]
- Kellner, RLL; Dettner, K. Differential efficacy of toxic pederin in deterring potential arthropod predators of Paederus (Coleoptera: Staphylinidae) offspring. Oecologia 1996, 107, 293–300. [Google Scholar]
- Sakemi, S; Ichiba, T; Kohmoto, S; Saucy, G. Isolation and structure elucidation of onnamide A, a new bioactive metabolite of a marine sponge, Theonella sp. J Am Chem Soc 1988, 110, 4851–4853. [Google Scholar]
- Matsunaga, S; Fusetani, N; Nakao, Y. Eight new cytotoxic metabolites closely related to onnamide A from two marine sponges of the genus Theonella. Tetrahedron 1992, 48, 8369–8376. [Google Scholar]
- Kobayashi, J; Itagaki, F; Shigemori, H; Sasaki, T. Three new onnamide congeners from the Okinawan marine sponge Theonella sp. J Nat Prod 1993, 56, 976–981. [Google Scholar]
- Vuong, D; Capon, RJ; Lacey, E; Gill, JH; Heiland, K; Friedel, T. Onnamide F: A new nematocide from a Southern Australian marine sponge Trachycladus laevispirulifer. J Nat Prod 2001, 64, 640–642. [Google Scholar]
- Fusetani, N; Sugawara, T; Matsunaga, S. Theopederins A–E, potent antitumor metabolites from a marine sponge, Theonella sp. J Org Chem 1992, 57, 3828–3832. [Google Scholar]
- Tsukamoto, S; Matsunaga, S; Fusetani, N; Toh-e, A. Theopederins F-J: Five new antifungal and cytotoxic metabolites from the marine sponge Theonella swinhoei. Tetrahedron 1999, 55, 13697–13702. [Google Scholar]
- Paul, GK; Gunasekera, SP; Longley, RE; Pomponi, SA. Theopederins K and L. Highly potent cytotoxic metabolites from a marine sponge Discodermia species. J Nat Prod 2002, 65, 59–61. [Google Scholar]
- Burres, NS; Clement, JJ. Antitumor activity and mechanism of action of the novel marine natural products mycalamide-A and -B and onnamide. Cancer Res 1989, 49, 2935–2940. [Google Scholar]
- Carrasco, L; Fernandez-Puentes, C; Vazques, D. Antibiotics and compounds affecting translation by eukaryotic ribosomes. Specific enhancement of aminoacyl-tRNA binding by methylxanthines. Mol Cell Biochem 1976, 10, 97–122. [Google Scholar]
- Abell, AD; Blunt, JW; Foulds, GJ; Munro, MHG. Chemistry of the mycalamides: antiviral and antitumor compounds from a New Zealand marine sponge. Part 6. The synthesis and testing of analogues of the C(7)–C(10) fragment. J Chem Soc Perkin Trans 1 1997, 1647–1654. [Google Scholar]
- Gürel, G; Blaha, G; Steitz, TA; Moore, PB. Structures of triacetyloleandomycin and mycalamide A bind to the large ribosomal subunit of Haloarcula marismortui. Antimicrob Agents Chemother 2009, 53, 5010–5014. [Google Scholar]
- Hong, CY; Kishi, Y. Total synthesis of mycalamides A and B. J Org Chem 1990, 55, 4242–4245. [Google Scholar]
- Roush, WR; Pfeifer, LA. Total synthesis of mycalamide A and 7-epi-mycalamide A. Org Lett 2000, 2, 859–862. [Google Scholar]
- Sohn, J-H; Waizumi, N; Zhong, M; Rawal, VH. Total synthesis of mycalamide A. J Am Chem Soc 2005, 127, 7290–7291. [Google Scholar]
- Kagawa, N; Ihara, M; Toyota, M. Convergent total synthesis of (+)-mycalamide A. J Org Chem 2006, 71, 6796–6805. [Google Scholar]
- Kagawa, N; Ihara, M; Toyota, M. Total synthesis of (+)-mycalamide A. Org Lett 2006, 8, 875–878. [Google Scholar]
- Thompson, AM; Blunt, JW; Munro, MHG; Perry, NB; Pannell, LK. Chemistry of the mycalamides, antiviral and antitumour compounds from a marine sponge. Part 3. Acyl, alkyl and silyl derivatives. J Chem Soc, Perkin Trans 1 1992, 1335–1342. [Google Scholar]
- Thompson, AM; Blunt, JW; Munro, MHG; Clark, BM. Chemistry of the mycalamides, antiviral and antitumour compounds from a marine sponge. Part 4. Reactions of mycalamide A and alkyl derivatives with basic nucleophiles. J Chem Soc, Perkin Trans 1 1994, 1025–1031. [Google Scholar]
- Thompson, AM; Blunt, JW; Munro, MHG; Perry, NB. Chemistry of the mycalamides, antiviral and antitumor compounds from a marine sponge. Part 5. Acid-catalysed hydrolysis and acetal exchange, double bond additions and oxidation reactions. J Chem Soc, Perkin Trans 1 1995, 1233–1242. [Google Scholar]
- Fukui, H; Tsuchiya, Y; Fujita, K; Nakagawa, T; Koshino, K; Nakata, T. Synthesis and biological activity of artificial analogs of mycalamide A. Bioorg Med Chem Lett 1997, 7, 2081–2086. [Google Scholar]
- Richter, A; Kocienski, P; Raubo, P; Davies, D. The in vitro biological activities of synthetic 18-O-methyl mycalamide B, 10-epi-18-O-methyl mycalamide B and pederin. Anticancer Drug Des 1997, 12, 217–227. [Google Scholar]
- Northcote, PT; Blunt, JW; Munro, MHG. Pateamine: a potent cytotoxin from the New Zealand marine sponge, Mycale sp. Tetrahedron Lett 1991, 32, 6411–6414. [Google Scholar]
- Romo, D; Rzasa, RM; Shea, HA; Park, K; Langenhan, JM; Sun, L; Akhiezer, A; Liu, JO. Total synthesis and immunosuppressive activity of (−)-pateamine A and related compounds: Implementation of a β-lactam-based macrocyclization. J Am Chem Soc 1998, 120, 12237–12254. [Google Scholar]
- Licitra, EJ; Liu, JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc Nat Acad Sci USA 1996, 93, 12817–12821. [Google Scholar]
- Bordeleau, ME; Matthews, J; Wojnar, J; Lindqvist, L; Novac, O; Jankowsky, E; Sonenberg, N; Northcote, P; Teesdale-Spittle, P; Pelletier, J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc Nat Acad Sciences USA 2005, 102, 10460–10465. [Google Scholar]
- Low, WK; Dang, Y; Schneider-Poetsch, T; Shi, Z; Choi, NS; Merrick, WC; Romo, D; Liu, JO. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell 2005, 20, 709–722. [Google Scholar]
- Low, WK; Dang, Y; Schneider-Poetsch, T; Shi, Z; Choi, NS; Rzasa, RM; Shea, HA; Li, S; Park, K; Ma, G; Romo, D; Liu, JO. Isolation and identification of eukaryotic initiation factor 4A as a molecular target for the marine natural product Pateamine A. Meth Enzymol 2007, 431, 303–324. [Google Scholar]
- Dang, Y; Low, WK; Xu, J; Gehring, NH; Dietz, HC; Romo, D; Liu, JO. Inhibition of nonsense-mediated mRNA decay by the natural product pateamine A through eukaryotic initiation factor 4AIII. J Biol Chem 2009, 284, 23613–23621. [Google Scholar]
- West, LM; Northcote, PT; Battershill, CN. Peloruside A: A potent cytotoxic macrolide isolated from the New Zealand marine sponge Mycale sp. J Org Chem 2000, 65, 445–449. [Google Scholar]
- Hood, KA; West, LM; Rouwé, B; Northcote, PT; Berridge, MV; Wakefield, StJ; Miller, JH. Peloruside A, a novel anti-mitotic agent with paclitaxel-like microtubule-stabilizing activity. Cancer Res 2002, 62, 3356–3360. [Google Scholar]
- Rowinsky, EK; Eisenhauer, EA; Chaudhry, V; Arbuck, SG; Donehower, RC. Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol 1993, 20, 1–15. [Google Scholar]
- Horwitz, SB. Personal recollections on the early development of Taxol. J Nat Prod 2004, 67, 136–138. [Google Scholar]
- Díaz, JF; Andreu, JM. Assembly of purified GDP-tubulin into microtubules induced by Taxol and Taxotere: Reversibility, ligand stoichiometry, and competition. Biochemistry 1993, 32, 2747–2755. [Google Scholar]
- Gaitanos, TN; Buey, RM; Díaz, JF; Northcote, PT; Teesdale-Spittle, P; Andreu, JM; Miller, JH. Peloruside A does not bind to the taxoid site on β-tubulin and retains its activity in multidrug resistant cell lines. Cancer Res 2004, 64, 5063–5067. [Google Scholar]
- Kowalski, RJ; Giannakakou, P; Gunasekera, SP; Longley, RE; Day, BW; Hamel, E. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (Taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paclitaxel-resistant cells. Mol Pharmacol 1997, 52, 613–622. [Google Scholar]
- Yvon, AMC; Wadsworth, P; Jordan, MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell 1999, 10, 947–959. [Google Scholar]
- Kamath, K; Jordan, MA. Suppression of microtubule dynamics by epothilone B is associated with mitotic arrest. Cancer Res 2003, 63, 6026–6031. [Google Scholar]
- Honore, S; Kamath, K; Braguer, D; Wilson, L; Briand, C; Jordan, MA. Suppression of microtubule dynamics by discodermolide by a novel mechanism is associated with mitotic arrest and inhibition of tumor cell proliferation. Mol Cancer Ther 2003, 2, 1303–1311. [Google Scholar]
- Honore, S; Kamath, K; Braguer, D; Horwitz, SB; Wilson, L; Briand, C; Jordan, MA. Synergistic suppression of microtubule dynamics by discodermolide and paclitaxel in non-small cell lung carcinoma cells. Cancer Res 2004, 64, 4957–4964. [Google Scholar]
- Chan, A; Andreae, P; Northcote, PT; Miller, JH. Peloruside A inhibits microtubule dynamics in a breast cancer cell line MCF7. Invest New Drugs 2010. [Google Scholar] [CrossRef]
- Wilmes, A; Rawson, P; Peng, L; McLauchlan, D; Northcote, PT; Jordan, TW; Miller, JH. Effects of the microtubule stabilizing agent peloruside A on the proteome of HL-60 cells. Invest New Drugs 2010. [Google Scholar] [CrossRef]
- Hermeking, H; Eick, D. Mediation of c-Myc-induced apoptosis by p53. Science 1994, 265, 2091–2093. [Google Scholar]
- Pryor, DE; O’Brate, A; Bilcer, G; Díaz, JF; Wang, Y; Wang, Y; Kabaki, M; Jung, MK; Andreu, JM; Ghosh, AK; Giannakakou, P; Hamel, E. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 2002, 41, 9109–9115. [Google Scholar]
- Nogales, E; Wolf, SG; Khan, IA; Ludueña, RF; Downing, KH. Structure of tubulin at 6.5 Å and location of the taxol-binding site. Nature 1995, 375, 424–427. [Google Scholar]
- Downing, KH. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu Rev Cell Dev Biol 2000, 16, 89–111. [Google Scholar]
- Hamel, E; Day, BW; Miller, JH; Jung, MK; Northcote, PT; Ghosh, AK; Curran, DP; Cushman, M; Nicolaou, KC; Paterson, I; Sorensen, EJ. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol Pharmacol 2006, 70, 1555–1564. [Google Scholar]
- Wilmes, A; Bargh, K; Kelly, C; Northcote, PT; Miller, JH. Peloruside A synergizes with other microtubule stabilizing agents in cultured cancer cell lines. Mol Pharm 2007, 4, 269–280. [Google Scholar]
- Huzil, JT; Chik, JK; Slysz, GW; Freedman, H; Tuszynski, J; Taylor, RE; Sackett, DL; Schriemer, DC. A unique mode of microtubule stabilization induced by peloruside A. J Mol Biol 2008, 378, 1016–1030. [Google Scholar]
- Pineda, O; Farràs, J; Maccari, L; Manetti, F; Botta, M; Vilarrasa, J. Computational comparison of microtubule-stabilizing agents laulimalide and peloruside with taxol and colchicine. Bioorg Med Chem Lett 2004, 14, 4825–4829. [Google Scholar]
- Jiménez-Barbero, J; Canales, A; Northcote, PT; Buey, RM; Andreu, JM; Díaz, JF. NMR determination of the bioactive conformation of peloruside A bound to microtubules. J Am Chem Soc 2006, 128, 8757–8765. [Google Scholar]
- Barberis, A; Gunde, T; Berset, C; Audetat, S; Lüthi, U. Yeast as a screening tool. Drug Discov Today: Technol 2005, 2, 187–192. [Google Scholar]
- Bode, CJ; Gupta, ML, Jr; Reiff, EA; Suprenant, KA; Georg, GI; Himes, RH. Epothilone and paclitaxel: unexpected differences in promoting the assembly and stabilization of yeast microtubules. Biochemistry 2002, 41, 3870–3874. [Google Scholar]
- Gupta, ML, Jr; Bode, CL; Georg, GL; Himes, RH. Understanding tubulin-Taxol interactions: Mutations that impart Taxol binding to yeast tubulin. Proc Nat Acad Sci USA 2003, 100, 6394–6397. [Google Scholar]
- Tong, AHY; Evangelista, M; Parsons, AB; Xu, H; Bader, GD; Pagé, N; Robinson, M; Raghibizadeh, S; Hogue, CWV; Bussey, H; Andrews, B; Tyers, M; Boone, C. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2001, 294, 2364–2368. [Google Scholar]
- Giaever, G; Flaherty, P; Kumm, J; Proctor, M; Nislow, C; Jaramillo, DF; Chu, AM; Jordan, MI; Arkin, AP; Davis, RW. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proc Nat Acad Sci USA 2004, 101, 793–798. [Google Scholar]
- Vanhecke, D; Janitz, M. High-throughput gene silencing using cell arrays. Oncogene 2004, 23, 8353–8358. [Google Scholar]
- Whitehurst, AW; Bodemann, BO; Cardenas, J; Ferguson, D; Girard, L; Peyton, M; Minna, JD; Michnoff, C; Hao, W; Roth, MG; Xie, XJ; White, MA. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar]
- Schadt, EE; Friend, SH; Shaywitz, DA. A network view of disease and compound screening. Nat Rev Drug Discov 2009, 8, 286–295. [Google Scholar]
- Costanzo, M; Baryshnikova, A; Bellay, J; Kim, Y; Spear, ED; Sevier, CS; Ding, H; Koh, JLY; Toufighi, K; Mostafavi, S; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar]
- Khrapunovich-Baine, M; Menon, V; Verdier-Pinard, P; Smith, AB, III; Angeletti, RH; Fiser, A; Horwitz, SB; Xiao, H. Distinct pose of discodermolide in taxol binding pocket drives a complementary mode of microtubule stabilization. Biochemistry 2009, 48, 11664–11677. [Google Scholar]
- Xiao, H; Verdier-Pinard, P; Fernandez-Fuentes, N; Burd, B; Angeletti, R; Fiser, A; Horwitz, SB; Orr, GA. Insights into the mechanism of microtubule stabilization by Taxol. Proc Nat Acad Sci USA 2006, 103, 10166–10173. [Google Scholar]
- Singh, AJ; Xu, CX; Xu, X; West, LM; Wilmes, A; Chan, A; Hamel, E; Miller, JH; Northcote, PT; Ghosh, AK. Peloruside B, a potent antitumor macrolide from the New Zealand marine sponge Mycale hentscheli: Isolation, structure, total synthesis and bioactivity. J Org Chem 2010, 75, 2–10. [Google Scholar]
- Northcote, PT; Miller, JH; West, LM; Hood, KA. Bioactive compound (Peloruside A). US Patent Number US 6,790,862 B2, 14 September 2004. [Google Scholar]
- Meyer, C; Ferguson, D; Krauth, M; Wick, M; Northcote, P. RTA 301 (peloruside): a novel microtubule stabilizer with potent in vivo activity against lung cancer and resistant breast cancer. 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics, Prague, Czech Republic, November 7–10, 2006; Abstr No. 639. Available online: http://www.sciencedirect.com/science/article/B75GT-4MMF4DM-ST/2/901656de391d50b1ced2b7ad843d922a. [CrossRef]
- Crume, KP; O’Sullivan, D; Miller, JH; Northcote, PT; La Flamme, AC. Delaying the onset of experimental autoimmune encephalomyelitis with the microtubule-stabilizing compounds, paclitaxel and peloruside A. J Leukocyte Biol 2009, 86, 949–958. [Google Scholar]
- Brunden, KR; Trojanowski, JQ; Lee, VM-Y. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov 2009, 8, 783–793. [Google Scholar]
- Liao, X; Wu, Y; De Brabander, JK. Total synthesis of peloruside A. Angew Chem Int Ed 2003, 42, 1648–1652. [Google Scholar]
- Jin, M; Taylor, RE. Total synthesis of (+)-peloruside A. Org Lett 2005, 7, 1303–1305. [Google Scholar]
- Ghosh, AK; Xu, X; Kim, JH; Xu, CX. Enantioselective total synthesis of peloruside A: a potent microtubule stabilizer. Org Lett 2008, 10, 1001–1004. [Google Scholar]
- Evans, DA; Welch, DS; Speed, AW; Moniz, GA; Reichelt, A; Ho, S. An aldol-based synthesis of (+)-peloruside a, a potent microtubule stabilizing agent. J Am Chem Soc 2009, 131, 3840–3841. [Google Scholar]
- Williams, DR; Nag, PP; Zorn, N. Strategies for the synthesis of the novel antitumor agent peloruside A. Curr Opin Drug Discov Devel 2008, 11, 251–271. [Google Scholar]
- Uenishi, J; Iwamoto, T; Tanaka, J. Total synthesis of (−)-zampanolide and questionable existence of (−)-dactylolide as the elusive biosynthetic precursor of (−)-zampanolide in an Okinawan sponge. Org Lett 2009, 11, 3262–3265. [Google Scholar]
- Tanaka, J; Higa, T. Zampanolide, a new cytotoxic macrolide from a marine sponge. Tetrahedron Lett 1996, 37, 5535–5538. [Google Scholar]
- Smith, AB, III; Safonov, IG; Corbett, RM. Total syntheses of (+)-zampanolide and (+)-dactylolide exploiting a unified strategy. J Am Chem Soc 2002, 124, 11102–11113. [Google Scholar]
- Field, JJ; Singh, AJ; Kanakkanthara, A; Halafihi, T; Northcote, PT; Miller, JH. Microtubule-stabilizing activity of Zampanolide, a potent macrolide isolated from the Tongan marine sponge Cacospongia mycofijiensis. J Med Chem 2009, 52, 7328–7332. [Google Scholar]
- Smith, AB, III; Safonov, IG; Corbett, RM. Total synthesis of (+)-zampanolide. J Am Chem Soc 2001, 123, 12426–12427. [Google Scholar]
- Ding, F; Jennings, MP. Total synthesis of (−)-dactylolide and formal synthesis of (−)-zampanolide via target oriented β-C-glycoside formation. J Org Chem 2008, 73, 5965–5976. [Google Scholar]
- Ding, F; Jennings, MP. An expedient total synthesis of (−)-dactylolide and formal synthesis of (−)-zampanolide. Org Lett 2005, 7, 2321–2324. [Google Scholar]
- Hoye, TR; Hu, MJ. Macrolactonization via Ti(IV)-mediated epoxy-acid coupling: a total synthesis of (−)-dactylolide [and zampanolide]. J Am Chem Soc 2003, 125, 9576–9577. [Google Scholar]
- Jennings, MP. Total Synthesis of (−)-dactylolide and formal synthesis of (−)zampanolide. Lett Org Chem 2006, 3, 78–81. [Google Scholar]





© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miller, J.H.; Singh, A.J.; Northcote, P.T. Microtubule-Stabilizing Drugs from Marine Sponges: Focus on Peloruside A and Zampanolide. Mar. Drugs 2010, 8, 1059-1079. https://doi.org/10.3390/md8041059
Miller JH, Singh AJ, Northcote PT. Microtubule-Stabilizing Drugs from Marine Sponges: Focus on Peloruside A and Zampanolide. Marine Drugs. 2010; 8(4):1059-1079. https://doi.org/10.3390/md8041059
Chicago/Turabian StyleMiller, John H., A. Jonathan Singh, and Peter T. Northcote. 2010. "Microtubule-Stabilizing Drugs from Marine Sponges: Focus on Peloruside A and Zampanolide" Marine Drugs 8, no. 4: 1059-1079. https://doi.org/10.3390/md8041059
APA StyleMiller, J. H., Singh, A. J., & Northcote, P. T. (2010). Microtubule-Stabilizing Drugs from Marine Sponges: Focus on Peloruside A and Zampanolide. Marine Drugs, 8(4), 1059-1079. https://doi.org/10.3390/md8041059