Characterization of Histone Genes from the Bivalve Lucina Pectinata

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological
2.2. DNA and RNA Isolation
2.3. Whole Genome Sequencing
2.4. RNA Sequencing
2.5. Sequence Similarity Searches and Validation of Semiconductor Sequencing Results
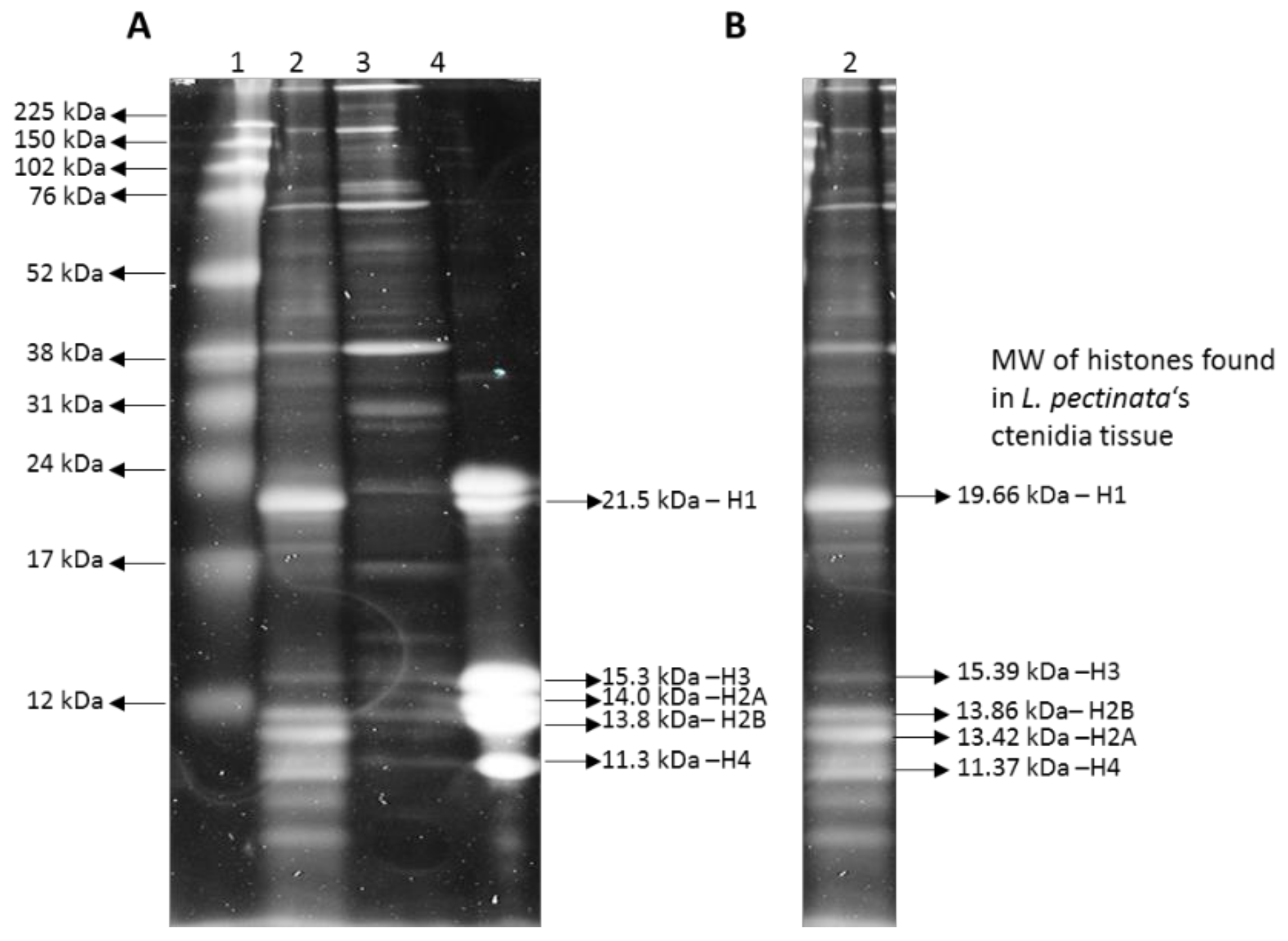
2.6. Histone Protein Extraction and Electrophoresis
2.7. Selection of Sequences from Different Species Related To The Histone H1_A Protein Sequence
2.8. Multiple Sequence Alignment of Histone H1 Proteins from Various Species and Phylogenetic Analysis of Histone H1 Proteins
3. Results
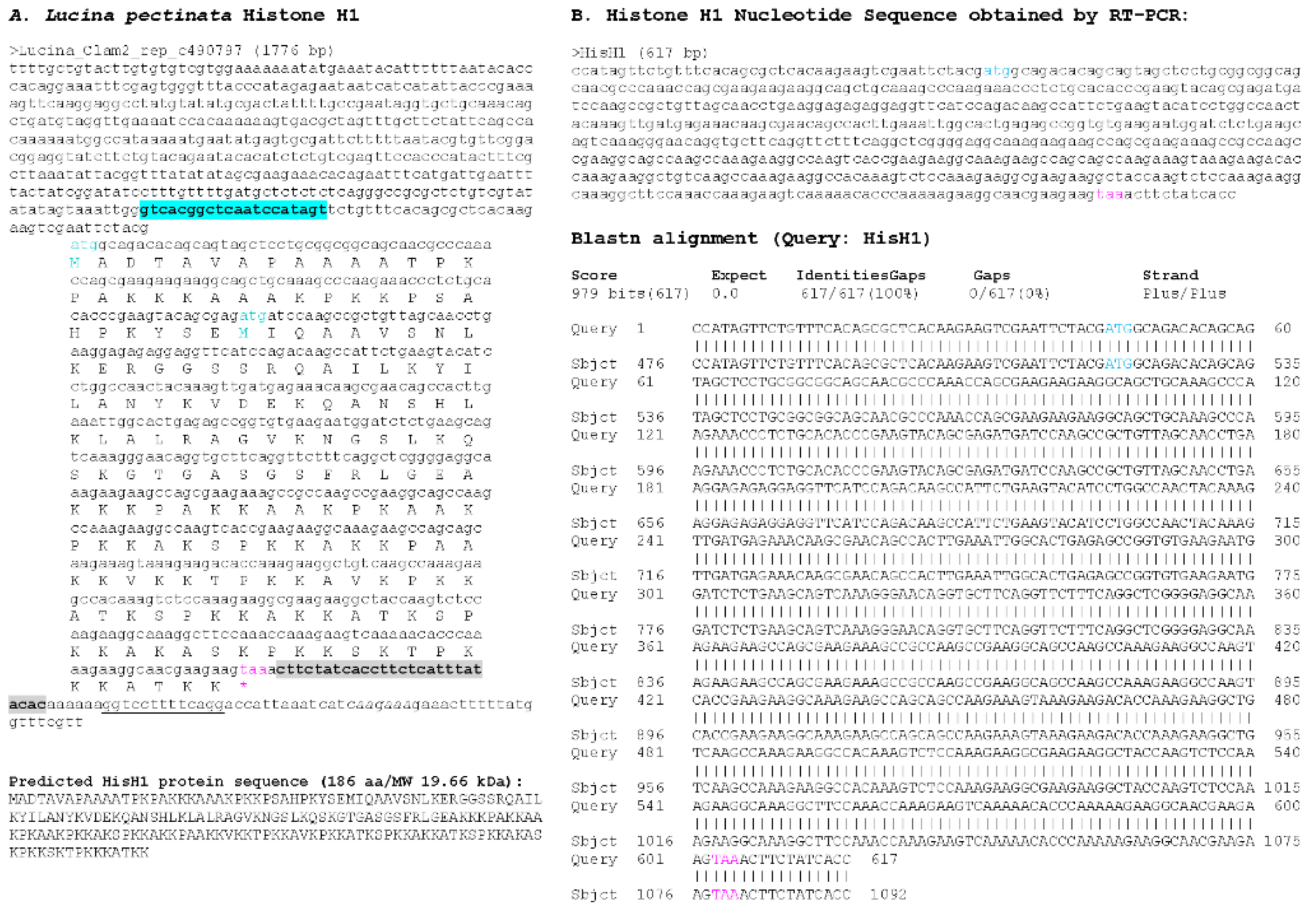
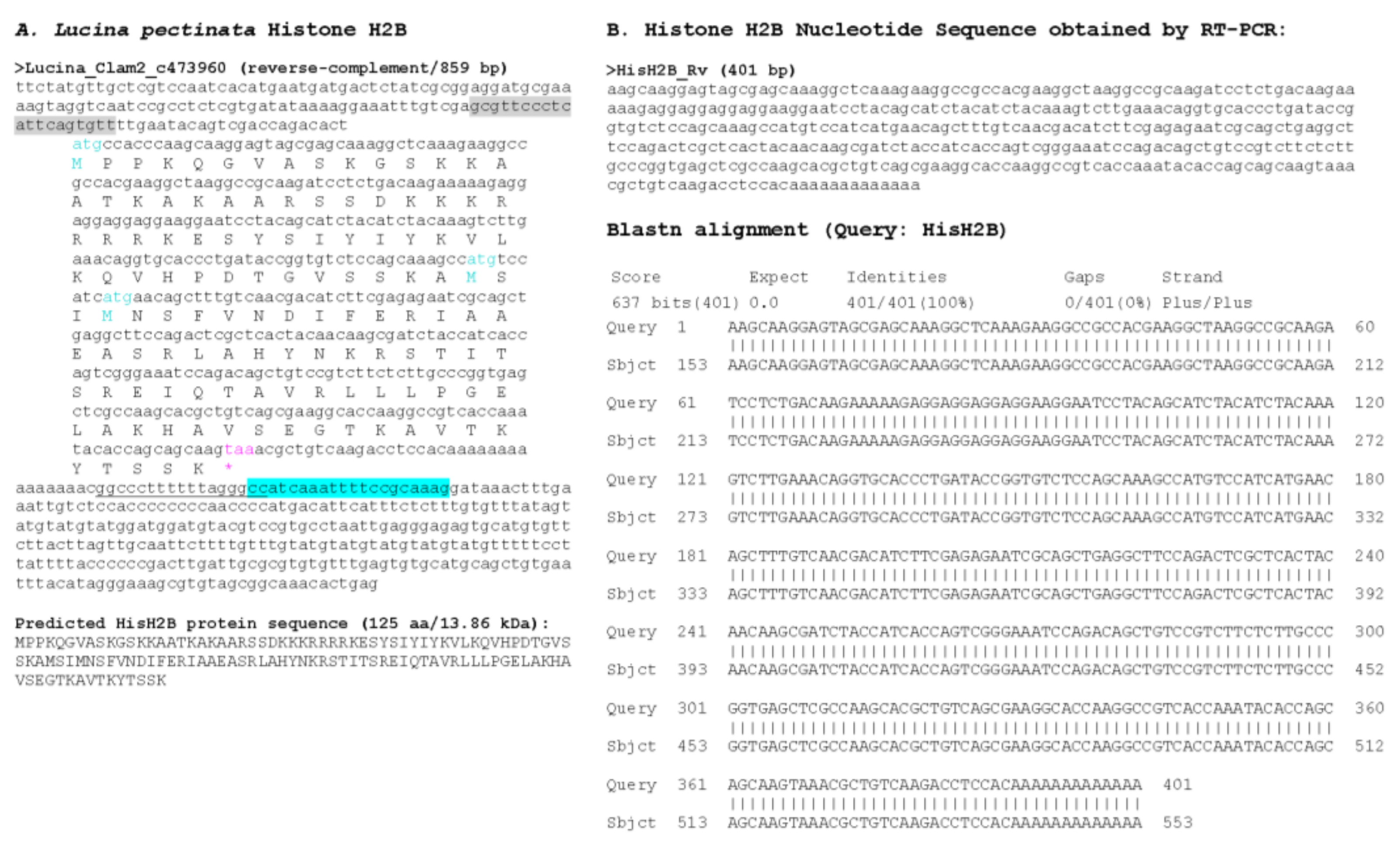
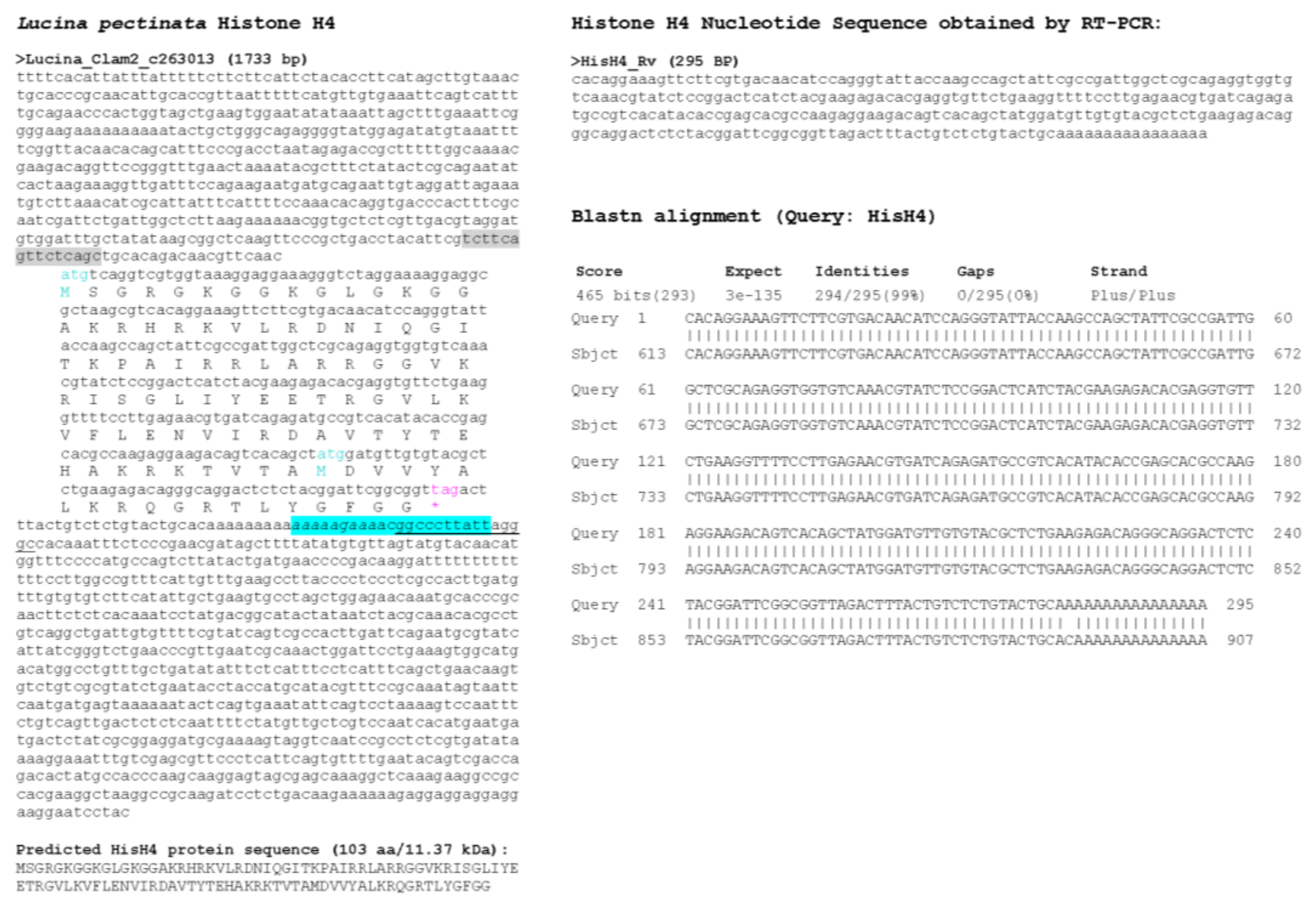
3.1. Sequence Similarity Searches Identify Lucina Pectinata Histone Genes in Genome Assembly
3.2. Multiple Sequence Alignment of Histone H1_A Variant Sequence Shows Conservation of Amino Acids Among Species
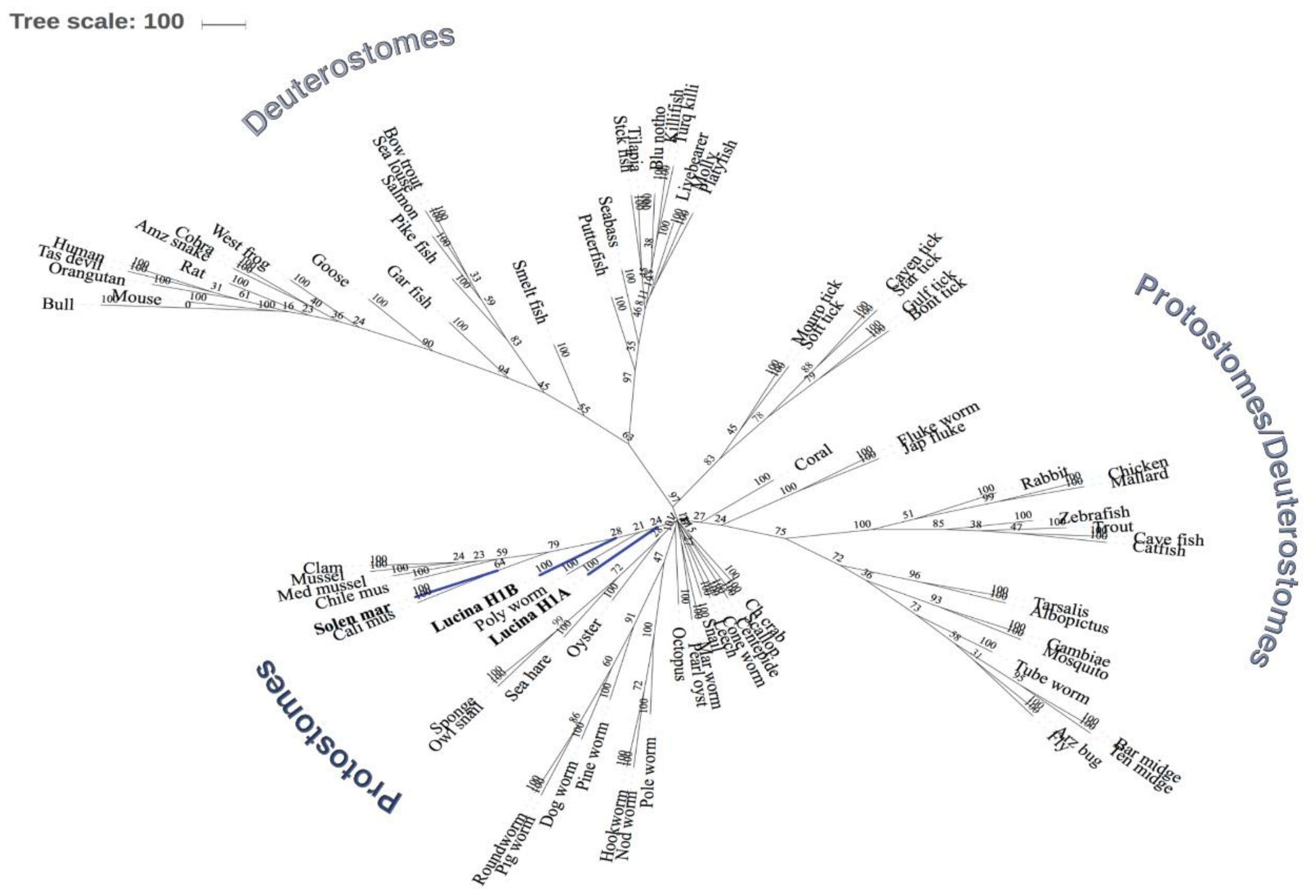
3.3. Phylogenetic Analysis of Lucina Pectinata Histone H1 Shows a Common Ancestor Among Other Histone H1 Proteins from Mollusks and Other Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- González-Romero, R.; Ausió, J.; Méndez, J.; Eirín-López, J. Histone genes of the razor clam Solen marginatus unveil new aspects of linker histone evolution in protostomes. Genome 2009, 52, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Olins, A.L.; Olins, D.E. Spheroid chromatin units (v bodies). Science 1974, 183, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Ponte, I.; Vila, R.; Suau, P. Sequence complexity of histone H1 subtypes. Mol. Biol. Evol. 2003, 20, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Thoma, F.; Koller, T. Influence of histone H1 on chromatin structure. Cell 1997, 12, 101–110. [Google Scholar] [CrossRef]
- Szerlong, H.J.; Hansen, J.C. Nucleosome distribution and linker DNA: Connecting nuclear function to dynamic chromatin structure. Biochem. Cell Biol. 2011, 89, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Eirín-López, J.; Fernanda, M.; González-Tizón, A.; Martínez, A.; Méndez, J. Birth-and-Death Evolution with Strong Purifying Selection in the Histone H1 Multigene Family and the Origin of orphon H1 Genes. Mol. Biol. Evol. 2004, 21, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Montes-Rodríguez, I.M.; Cadilla, C.L.; González-Méndez, R.; López-Garriga, J.; Ropelewski, A. De Novo Assembly of Lucina pectinata Genome using Ion Torrent Reads. In Proceedings of the Practice and Experience in Advanced Research Computing 2017 Proceedings (PEARC17), New Orleans, LA, USA, 9–13 July 2017. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Krebs, S.; Fischaleck, M.; Blum, H. A simple and loss-free method to remove TRIzol contaminations from minute RNA samples. Anal. Biochem. 2009, 387, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Church, D.M.; Federhen, S.; Lash, A.E.; Madden, T.L.; Pontius, J.U.; Schuler, G.D.; Schriml, L.M.; Sequeira, E.; Tatusova, T.A.; et al. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003, 31, 28–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Research. [CrossRef] [PubMed]
- Thomas, J.O.; Kornberg, R.D. An octamer of histones in chromatin and free in solution. Proc. Natl. Acad. Sci. USA 1975, 72, 2626–2630. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996, 266, 418–427. [Google Scholar] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Van Holde, K.A. Chapter 4: The Proteins of Chromatin. I. Histones. In Chromatin; Richi, A., Ed.; Springer-Verlag: New York, NY, USA, 1988; pp. 69–168. [Google Scholar]
- Maxson, R.; Cohn, R.; Kedes, L.; Mohun, T. Expression and Organization of Histone Genes. Ann. Rev. Genet. 1983, 17, 239–277. [Google Scholar] [CrossRef] [PubMed]
- Eirin-López, J.M.; Ruiz, M.F.; Gonález-Tizón, A.M.; Martínez, A.; Ausió, J.; Sánchez, L.; Méndez, J. Common evolutionary origin and birth-and-death process in the replication-independent histone H1 isoforms from vertebrate and invertebrate genomes. J. Mol. Evol. 2005, 61, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. De novo genome assembly: What every biologist should know. Nat. Methods 2012, 9, 333–337. [Google Scholar] [CrossRef]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2013, 13, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Oki, N.; Yano, K.; Okumoto, Y.; Tsukiyama, T.; Teraishi, M.; Tanisaka, T. A genome-wide view of miniature inverted-repeat transposable elements (MITEs) in rice, Oryza sativa ssp. japonica. Genes Genet. Syst. 2008, 83, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.B.; Chodchoy, N.; Liu, T.J.; Marzluff, W.F. Introns in histone genes alter the distribution of 3′ ends. Nucleic Acids Res. 1990, 18, 3161–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Romero, R.; Rivera-Casas, C.; Frehlick, L.J.; Méndez, J.; Ausió, J.; Eirín-López, J.M. Histone H2A (H2A.X and H2A.Z) variants in molluscs: Molecular characterization and potential implications for chromatin dynamics. PLoS ONE 2012, 7, e30006. [Google Scholar] [CrossRef] [PubMed]
- Mariño-Ramírez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating scientific discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]
- Nystrom, N.; Welling, J.; Blood, P.; Goh, E.L. Blacklight: Coherent Shared Memory for Enabling Science. In Contemporary High Performance Computing: From Petascale toward Exascale, CRC 2013; Vetter, J.S., Ed.; Chapman and Hall: London, UK, 2013; pp. 431–450. [Google Scholar]
- Nystrom, N.A.; Levine, M.J.; Roskies, R.Z.; Scott, J. Bridges: A uniquely flexible HPC resource for new communities and data analytics. In Proceedings of the 2015 XSEDE Conference, Scientific Advancements Enabled by Enhanced Cyberinfrastructure, St. Louis, MO, USA, 26−30 July 2015; p. 30. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Sequence | Forward Primer | Reverse Primer |
|---|---|---|
| Histone H1_A | 5′-GTCACGGCTCAATCCATAGT-3′ | 5′-GTGTGATAAATGAGAAGGTGATAGAAG-3′ |
| Histone H2A | 5′-GGCGATGCCTCATATTCTTT-3′ | 5′-AAAAAGGGCCGTTGAAAGTT-3′ |
| Histone H2B | 5′-CTTTGCGGAAAATTTGATGG-3′ | 5′-GCGTTCCCTCATTCAGTGTT-3′ |
| Histone H3 | 5′-AAAAAGGGCCGTTATAGTGAG-3′ | 5′-ACCAATCAGCGTTGTTTTCC-3′ |
| Histone H4 | 5′-AATAAGGGCCGTTTTCTTTTT-3′ | 5′-CGGGTTCTTCAGTTCTCAGC-3′ |
| Query Seq-id | Subject Seq-id | % Identity | Alignment Length | Number of Mismatches | # of Gaps | Start Query | End Query | Start subject | End Subject | E-value | Bit score |
|---|---|---|---|---|---|---|---|---|---|---|---|
| L.pectinataH3 | Lucina_Clam2_rep_c512804 | 99.7 | 328 | 1 | 0 | 1 | 328 | 139 | 466 | 9.00E-173 | 589 |
| S.margina-tus.H3 | Lucina_Clam2_rep_c512804 | 82.75 | 458 | 74 | 2 | 200 | 652 | 115 | 572 | 2.00E-135 | 466 |
| S.margina-tus.H1 | Lucina_Clam2_c212178 | 68.98 | 432 | 92 | 12 | 251 | 670 | 1899 | 1498 | 1.00E-42 | 159 |
| S.margina-tus.H1 | Lucina_Clam2_rep_c490797 | 68.98 | 432 | 92 | 12 | 251 | 670 | 520 | 921 | 1.00E-42 | 159 |
| S.margina-tus.H1 | Lucina_Clam2_rep_c490798 | 68.98 | 432 | 92 | 12 | 251 | 670 | 420 | 821 | 5.00E-43 | 159 |
| S.margina-tus.H2B | Lucina_Clam2_c473960 | 76.92 | 429 | 89 | 5 | 171 | 593 | 690 | 266 | 9.00E-90 | 315 |
| S.margina-tus.H3 | Lucina_Clam2_rep_c512805 | 81.37 | 424 | 73 | 3 | 234 | 652 | 1 | 423 | 9.00E-116 | 401 |
| S.margina-tus.H2A | Lucina_Clam2_c411227 | 76.47 | 391 | 88 | 2 | 146 | 532 | 694 | 1084 | 6.00E-81 | 286 |
| S.margina-tus.H2A | Lucina_Clam2_c263014 | 76.67 | 390 | 88 | 1 | 146 | 532 | 52 | 441 | 1.00E-82 | 291 |
| S.margina-tus.H2A | Lucina_Clam2_c46738 | 68.51 | 362 | 107 | 2 | 151 | 507 | 241 | 600 | 6.00E-36 | 136 |
| S.margina-tus.H2A | Lucina_Clam2_c75853 | 67.04 | 361 | 105 | 5 | 151 | 505 | 857 | 505 | 8.00E-27 | 105 |
| S.margina-tus.H2B | Lucina_Clam2_c411226 | 79.2 | 327 | 65 | 1 | 171 | 497 | 417 | 740 | 4.00E-80 | 282 |
| S.margina-tus.H4 | Lucina_Clam2_c263013 | 79.62 | 319 | 65 | 0 | 147 | 465 | 555 | 873 | 9.00E-80 | 282 |
| S.margina-tus.H4 | Lucina_Clam2_rep_c482374 | 79.62 | 319 | 65 | 0 | 147 | 465 | 5 | 323 | 7.00E-80 | 282 |
| S.margina-tus.H4 | Lucina_Clam2_rep_c482375 | 79.31 | 319 | 65 | 1 | 147 | 465 | 46 | 363 | 4.00E-78 | 275 |
| S.margina-tus.H1 | Lucina_Clam2_c353214 | 69.18 | 305 | 81 | 7 | 302 | 601 | 317 | 613 | 3.00E-27 | 107 |
| Solen.marginatus.H2A | Lucina_Clam2_rep_c481900 | 76.6 | 282 | 65 | 1 | 146 | 427 | 281 | 1 | 1.00E-57 | 208 |
| Solen.marginatus.H3 | Lucina_Clam2_rep_c482111 | 83.27 | 275 | 46 | 0 | 200 | 474 | 327 | 601 | 3.00E-82 | 289 |
| Solen.marginatus.H3 | Lucina_Clam2_c3893 | 74.35 | 269 | 65 | 2 | 200 | 466 | 1111 | 845 | 5.00E-45 | 168 |
| Solen.marginatus.H2A | Lucina_Clam2_c155385 | 72.08 | 265 | 73 | 1 | 143 | 407 | 722 | 985 | 1.00E-37 | 141 |
| Solen.marginatus.H1 | Lucina_Clam2_rep_c482183 | 69.61 | 204 | 35 | 7 | 473 | 670 | 11 | 193 | 2.00E-19 | 82.4 |
| Type of Histone | Protein Size1 | pI2 | MW3 | Solen Marginatus Protein Size3 | Solen Marginatus Protein pI2 | Solen Marginatus Protein MW3 |
|---|---|---|---|---|---|---|
| H1_A | 186 | 10.94 | 19,658 | 190 | 11.18 | 19,918 |
| H1_B | 179 | 11.29 | 18,750 | |||
| H3 | 136 | 11.27 | 15,388 | 136 | 11.47 | 15,521 |
| H4 | 103 | 11.36 | 11,367 | 103 | 11.36 | 11,441 |
| H2A_W | 135 | 10.12 | 14,687 | 125 | 10.90 | 13,435 |
| H2A_X | 126 | 10.90 | 13,415 | |||
| H2A_Y | 127 | 10.58 | 13,377 | |||
| H2A_Z | 125 | 10.62 | 13,379 | |||
| H2B | 125 | 10.52 | 13,855 | 124 | 10.71 | 13,837 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montes-Rodríguez, I.M.; Rodríguez-Pou, Y.; González-Méndez, R.R.; Lopez-Garriga, J.; Ropelewski, A.J.; Cadilla, C.L. Characterization of Histone Genes from the Bivalve Lucina Pectinata. Int. J. Environ. Res. Public Health 2018, 15, 2170. https://doi.org/10.3390/ijerph15102170
Montes-Rodríguez IM, Rodríguez-Pou Y, González-Méndez RR, Lopez-Garriga J, Ropelewski AJ, Cadilla CL. Characterization of Histone Genes from the Bivalve Lucina Pectinata. International Journal of Environmental Research and Public Health. 2018; 15(10):2170. https://doi.org/10.3390/ijerph15102170
Chicago/Turabian StyleMontes-Rodríguez, Ingrid M., Yesenia Rodríguez-Pou, Ricardo R. González-Méndez, Juan Lopez-Garriga, Alexander J. Ropelewski, and Carmen L. Cadilla. 2018. "Characterization of Histone Genes from the Bivalve Lucina Pectinata" International Journal of Environmental Research and Public Health 15, no. 10: 2170. https://doi.org/10.3390/ijerph15102170
APA StyleMontes-Rodríguez, I. M., Rodríguez-Pou, Y., González-Méndez, R. R., Lopez-Garriga, J., Ropelewski, A. J., & Cadilla, C. L. (2018). Characterization of Histone Genes from the Bivalve Lucina Pectinata. International Journal of Environmental Research and Public Health, 15(10), 2170. https://doi.org/10.3390/ijerph15102170