
Identification of Hub Genes in Different Stages of Colorectal Cancer through an Integrated Bioinformatics Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition of Microarray and RNA-Seq Data
2.2. Identification of Differentially Expressed Genes (DEGs) in GEO Microarray Datasets
2.3. Identification of Differentially Expressed Genes (DEGs) in RNA-Seq TCGA Datasets
2.4. Integration of Ranked Lists of Differentially Expressed Genes (DEGs) in Groups G1, G2, and G3
2.5. Functional and Pathway Enrichment Analysis
2.6. Protein–Protein Interaction
2.7. Hub Gene Screening and Analysis
2.8. Identification of Oxidative Stress-Response and Apoptosis-Associated Genes in CRC with Oncomine
3. Results
3.1. Genes Differentially Expressed in Colorectal Adenoma (G1) Datasets
3.2. Genes Differentially Expressed in Colorectal Adenocarcinoma (G2) Datasets
3.3. Genes Differentially Expressed in Colorectal Carcinoma (G3) Datasets
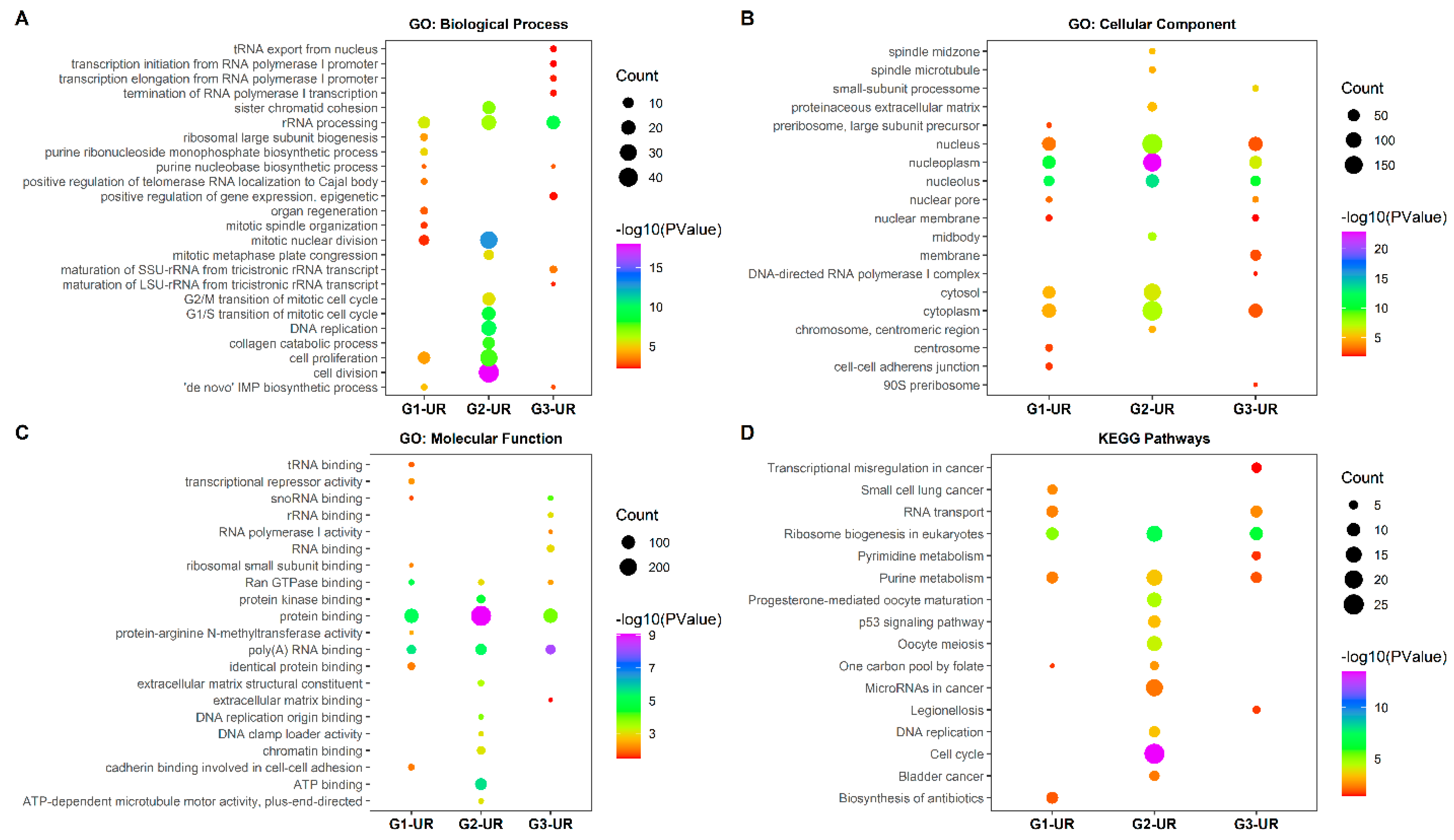
3.4. GO Term Enrichment Analysis
3.4.1. GO Terms and KEGG Pathway Enrichment Analysis of UR Genes
3.4.2. GO Terms and KEGG Pathway Enrichment Analysis of DR Genes
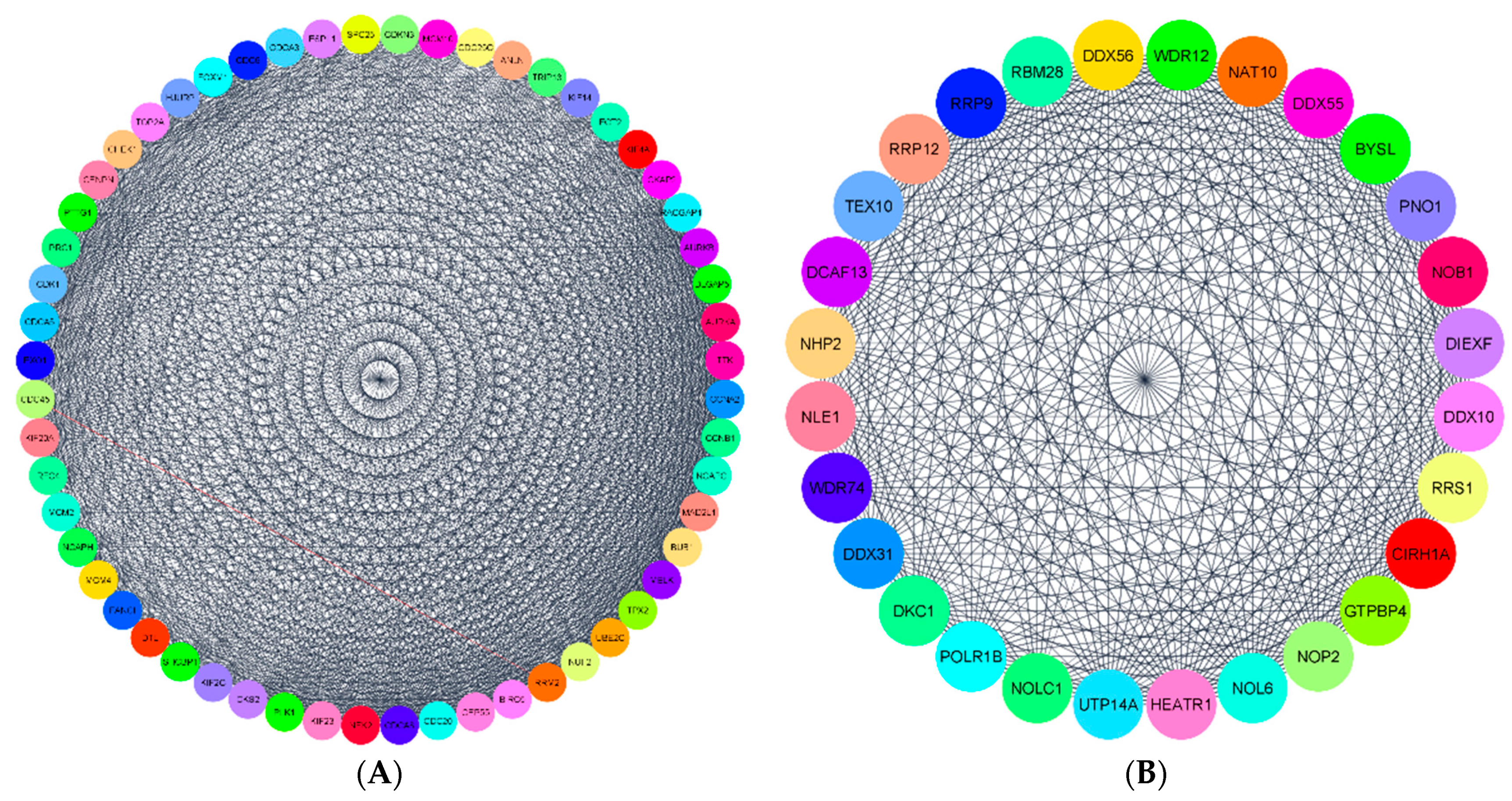
3.5. Protein-Protein Interaction (PPI) Network Construction and Module Selection for Key Genes
3.5.1. PPI Network Construction and Module Selection in G1, G2, and G3
3.5.2. Functional Enrichment of Hub Genes in KEGG Pathways
3.6. Analyzing DEGs in Oxidative Stress and Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, V.; Victorino, A.P.; Thuler, L.C.; Ferreira, C.G. Colorectal Cancer: Epidemiology, Disease Mechanisms and Interventions to Reduce Onset and Mortality. Clin. Colorectal Cancer 2016, 15, 195–203. [Google Scholar] [CrossRef]
- Ding, X.; Duan, H.; Luo, H. Identification of Core Gene Expression Signature and Key Pathways in Colorectal Cancer. Front. Genet. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brody, H. Colorectal cancer. Nature 2015, 521, S1. [Google Scholar] [CrossRef]
- Thorsteinsson, M.; Jess, P. The clinical significance of circulating tumor cells in non-metastatic colorectal cancer—A review. Eur. J. Surg. Oncol. 2011, 37, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Ma, Y.; Zhang, J.; Gu, J.; Jing, X.; Lu, S.; Fu, S.; Huo, J. Identification and Verification of Core Genes in Colorectal Cancer. BioMed Res. Int. 2020. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, J.; Wang, J.; Xu, M.; Zhang, Y.; Sun, P.; Liang, L. Identification Hub Genes in Colorectal Cancer by Integrating Weighted Gene Co-Expression Network Analysis and Clinical Validation in vivo and vitro. Front. Oncol. 2020. [Google Scholar] [CrossRef]
- Chen, Z.; Lin, Y.; Gao, J.; Lin, S.; Zheng, Y.; Liu, Y.; Chen, S.Q. Identification of key candidate genes for colorectal cancer by bioinformatics analysis. Oncol. Lett. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Momma, T.; Kono, K. Targeted therapy according to next generation sequencing-based panel sequencing. Fukushima J. Med. Sci. 2018. [Google Scholar] [CrossRef] [Green Version]
- Deshiere, A.; Berthet, N.; Lecouturier, F.; Gaudaire, D.; Hans, A. Molecular characterization of Equine Infectious Anemia Viruses using targeted sequence enrichment and next generation sequencing. Virology 2019. [Google Scholar] [CrossRef]
- Solé, X.; Crous-Bou, M.; Cordero, D.; Olivares, D.; Guinó, E.; Sanz-Pamplona, R.; Rodriguez-Moranta, F.; Sanjuan, X.; De Oca, J.; Salazar, R.; et al. Discovery and validation of new potential biomarkers for early detection of colon cancer. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Baloch, Z.; Ma, Y.; Wan, Z.; Huo, Y.; Li, F.; Zhao, Y. Identification of Potential Key Genes and Pathways in Early-Onset Colorectal Cancer through Bioinformatics Analysis. Cancer Control 2019. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, Z.; Shen, X.; Cui, X.; Guo, Y. Identification of novel biomarkers and small molecule drugs in human colorectal cancer by microarray and bioinformatics analysis. Mol. Genet. Genom. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Bao, Y.; Ma, M.; Yang, W. Identification of key candidate genes and pathways in colorectal cancer by integrated bioinformatical analysis. Int. J. Mol. Sci. 2017, 4, 722. [Google Scholar] [CrossRef] [Green Version]
- Sabates-Bellver, J.; Van Der Flier, L.G.; De Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzypczak, M.; Goryca, K.; Rubel, T.; Paziewska, A.; Mikula, M.; Jarosz, D.; Pachlewski, J.; Oledzki, J.; Ostrowsk, J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS ONE 2010. [Google Scholar] [CrossRef]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [Green Version]
- Gaedcke, J.; Grade, M.; Jung, K.; Camps, J.; Jo, P.; Emons, G.; Gehoff, A.; Sax, U.; Schirmer, M.; Becker, H.; et al. Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosom. Cancer 2010. [Google Scholar] [CrossRef] [Green Version]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013. [Google Scholar] [CrossRef] [Green Version]
- Vlachavas, E.I.; Pilalis, E.; Papadodima, O.; Koczan, D.; Willis, S.; Klippel, S.; Cheng, C.; Pan, L.; Sachpekidis, C.; Pintzas, A.; et al. Radiogenomic Analysis of F-18-Fluorodeoxyglucose Positron Emission Tomography and Gene Expression Data Elucidates the Epidemiological Complexity of Colorectal Cancer Landscape. Comput. Struct. Biotechnol. J. 2019. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Graudens, E.; Boulanger, V.; Mollard, C.; Mariage-Samson, R.; Barlet, X.; Grémy, G.; Couillault, C.; Lajémi, M.; Piatier-Tonneau, D.; Zaborski, P.; et al. Deciphering cellular states of innate tumor drug responses. Genome Biol. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamas, A.; Ishikawa, T.; Shimokawa, K.; Mogushi, K.; Iida, S.; Ishiguro, M.; Mizushima, H.; Tanaka, H.; Uetake, H.; Sugihara, K. Screening for epigenetically masked genes in colorectal cancer using 5-aza-2′-deoxycytidine, microarray and gene expression profile. Cancer Genom. Proteom. 2012. [Google Scholar]
- Dong, H.K.; Jeung, H.C.; Chan, H.P.; Seung, H.K.; Gui, Y.L.; Won, S.L.; Nam, K.K.; Hyun, C.C.; Sun, Y.R. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int. J. Cancer 2007. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef]
- Kolde, R.; Laur, S.; Adler, P.; Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 2012. [Google Scholar] [CrossRef] [Green Version]
- Nangraj, A.S.; Selvaraj, G.; Kaliamurthi, S.; Kaushik, A.C.; Cho, W.C.; Wei, D.Q. Integrated PPI- and WGCNA-Retrieval of Hub Gene Signatures Shared Between Barrett’s Esophagus and Esophageal Adenocarcinoma. Front. Pharmacol. 2020, 11, 1. [Google Scholar] [CrossRef]
- Hu, X.; Bao, M.; Huang, J.; Zhou, L.; Zheng, S. Identification and Validation of Novel Biomarkers for Diagnosis and Prognosis of Hepatocellular Carcinoma. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Hong, Y.; Won, J.; Lee, Y.; Lee, S.; Park, K.; Chang, K.T.; Hong, Y. Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J. Pineal Res. 2014. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.C.; Wallace, K.; Sandler, R.S.; Baron, J.A. Racial Disparities in Incidence of Young-Onset Colorectal Cancer and Patient Survival. Gastroenterology 2019. [Google Scholar] [CrossRef] [Green Version]
- Lech, G.; Słotwiński, R.; Słodkowski, M.; Krasnodębski, I.W. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Tsai, S.J. Genome-wide microarray analysis of gene expression profiling in major depression and antidepressant therapy. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016. [Google Scholar] [CrossRef]
- Wang, Z.Z.; Yang, J.; Jiang, B.H.; Di, J.B.; Gao, P.; Peng, L.; Su, X.Q. KIF14 promotes cell proliferation via activation of Akt and is directly targeted by miR-200c in colorectal cancer. Int. J. Oncol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Dwyer, M.; Okolotowicz, K.J.; Mercola, M.; Cashman, J.R. A novel inhibitor targets both WNT signaling and ATM/P53 in colorectal cancer. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, Y.Y.; Yang, X.F.; Shen, D.F.; Sun, H.Z.; Huang, K.Q.; Zheng, H.C. Effects and mechanism of STAT3 silencing on the growth and apoptosis of colorectal cancer cells. Oncol. Lett. 2018. [Google Scholar] [CrossRef]
- Gorbatenko, A.; Olesen, C.W.; Boedtkjer, E.; Pedersen, S.F. Regulation and roles of bicarbonate transporters in cancer. Front. Physiol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, Y.; Jiang, C.J.; Chen, Y.M.; Li, H.; Liu, Q.A. Calcium-Activated Chloride Channel A4 (CLCA4) Plays Inhibitory Roles in Invasion and Migration Through Suppressing Epithelial-Mesenchymal Transition via PI3K/AKT Signaling in Colorectal Cancer. Med. Sci. Monit. 2019. [Google Scholar] [CrossRef]
- Liu, G.; Wong, L.; Chua, H.N. Complex discovery from weighted PPI networks. Bioinformatics 2009. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.P.; Wang, L.P.; Wen, Y.Q.; Ren, X.Q.; Zuo, S.G. Identification of key genes for predicting colorectal cancer prognosis by integrated bioinformatics analysis. Oncol. Lett. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Kao, Y.; Zhang, C.; Sun, F.; Gong, Z.; Chen, J. Identification of Hub Genes Related to Carcinogenesis and Prognosis in Colorectal Cancer Based on Integrated Bioinformatics. Mediators Inflamm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Enzo, M.V.; Pucciarelli, S.; Bedin, C.; Pizzini, S.; Urso, E.D.L.; Nitti, D. Forkhead box gene (FOXO1A) the emerging role in colon cancer. Cancer Res. 2008, 68, 4147. [Google Scholar]
- Hong, B.S.; Cho, J.H.; Kim, H.; Choi, E.J.; Rho, S.; Kim, J.; Kim, J.H.; Choi, D.S.; Kim, Y.K.; Hwang, D.; et al. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genom. 2009. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Kao, T.P.; Huang, H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene 2008. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Nakayama, S.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Matsushima, T.; Torikoshi, Y.; Tanaka, S.; Yoshida, T.; Ishihara, H.; et al. Determination of the specific activity of CDK1 and CDK2 as a novel prognostic indicator for early breast cancer. Ann. Oncol. 2008. [Google Scholar] [CrossRef]
- Hansel, D.E.; Dhara, S.; Huang, R.C.C.; Ashfaq, R.; Deasel, M.; Shimada, Y.; Bernstein, H.S.; Harmon, J.; Brock, M.; Forastiere, A.; et al. CDC2/CDK1 expression in esophageal adenocarcinoma and precursor lesions serves as a diagnostic and cancer progression marker and potential novel drug target. Am. J. Surg. Pathol. 2005. [Google Scholar] [CrossRef]
- Wu, M.; Liu, Z.; Li, X.; Zhang, A.; Lin, D.; Li, N. Analysis of potential key genes in very early hepatocellular carcinoma. World J. Surg. Oncol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Piao, J.; Zhu, L.; Sun, J.; Li, N.; Dong, B.; Yang, Y.; Chen, L. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene 2019. [Google Scholar] [CrossRef]
- Chang, J.T.; Wang, H.M.; Chang, K.W.; Chen, W.H.; Wen, M.C.; Hsu, Y.M.; Yung, B.Y.M.; Chen, I.H.; Liao, C.T.; Hsieh, L.L.; et al. Identification of differentially expressed genes in oral squamous cell carcinoma (OSCC): Overexpression of NPM, CDK1 and NDRG1 and underexpression of CHES1. Int. J. Cancer 2005. [Google Scholar] [CrossRef]
- Lu, A.G.; Feng, H.; Wang, P.X.Z.; Han, D.P.; Chen, X.H.; Zheng, M.H. Emerging roles of the ribonucleotide reductase M2 in colorectal cancer and ultraviolet-induced DNA damage repair. World J. Gastroenterol. 2012. [Google Scholar] [CrossRef]
- Gan, Y.; Li, Y.; Li, T.; Shu, G.; Yin, G. CCNA2 acts as a novel biomarker in regulating the growth and apoptosis of colorectal cancer. Cancer Manag. Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Sano, B.; Nagata, T.; Kato, H.; Sugiyama, Y.; Kunieda, K.; Kimura, M.; Okano, Y.; Saji, S. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003. [Google Scholar] [CrossRef] [PubMed]
- Han, D.P.; Zhu, Q.L.; Cui, J.T.; Wang, P.X.; Qu, S.; Cao, Q.F.; Zong, Y.P.; Feng, B.; Zheng, M.H.; Lu, A.G. Polo-like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Med. Sci. Monit. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gali-Muhtasib, H.; Kuester, D.; Mawrin, C.; Bajbouj, K.; Diestel, A.; Ocker, M.; Habold, C.; Foltzer-Jourdainne, C.; Schoenfeld, P.; Peters, B.; et al. Thymoquinone triggers inactivation of the stress response pathway sensor CHEK1 and contributes to apoptosis in colorectal cancer cells. Cancer Res. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Ramakrishnan, S.K.; Weisz, K.; Triner, D.; Xie, L.; Attili, D.; Pant, A.; Győrffy, B.; Zhan, M.; Carter-Su, C.; et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab. 2016. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.; Shevchenko, A.; Mann, M.; Murray, A.W. Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J. Cell Biol. 1998. [Google Scholar] [CrossRef] [Green Version]
- Burum-Auensen, E.; DeAngelis, P.M.; Schjølberg, A.R.; Røislien, J.; Andersen, S.N.; Clausen, O.P.F. Spindle proteins Aurora A and BUB1B, but not Mad2, are aberrantly expressed in dysplastic mucosa of patients with longstanding ulcerative colitis. J. Clin. Pathol. 2007. [Google Scholar] [CrossRef] [Green Version]
- Sillars-Hardebol, A.H.; Carvalho, B.; Tijssen, M.; Beliën, J.A.M.; De Wit, M.; Delis-van Diemen, P.M.; Pontén, F.; Van De Wiel, M.A.; Fijneman, R.J.A.; Meijer, G.A. TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut 2012. [Google Scholar] [CrossRef] [Green Version]
- Ren, Q.; Jin, B. The clinical value and biological function of PTTG1 in colorectal cancer. Biomed. Pharmacother. 2017. [Google Scholar] [CrossRef]
- Ueki, T.; Nishidate, T.; Park, J.H.; Lin, M.L.; Shimo, A.; Hirata, K.; Nakamura, Y.; Katagiri, T. Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.W.; Chou, H.Y.E.; Liu, S.H.; Peng, S.Y.; Liu, C.L.; Hsu, H.C. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiaglia, E.; Selfe, J.; Hamdi, M.; Williamson, D.; Schaaf, G.; Fang, C.; Koster, J.; Summersgill, B.; Messahel, B.; Versteeg, R.; et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: An approach to identify candidate genes involved in tumor development. Genes Chromosom. Cancer 2009. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Rape, M. Regulated Degradation of Spindle Assembly Factors by the Anaphase-Promoting Complex. Mol. Cell 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.; Wei, Z.; Cui, H.; Zhang, W.; Wei, X.; Lu, Z.; Bai, X. NUSAP1 gene silencing inhibits cell proliferation, migration and invasion through inhibiting DNMT1 gene expression in human colorectal cancer. Exp. Cell Res. 2018. [Google Scholar] [CrossRef]
- Zhu, C.; Zhao, J.; Bibikova, M.; Leverson, J.D.; Bossy-Wetzel, E.; Fan, J.B.; Abraham, R.T.; Jiang, W. Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol. Biol. Cell 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, T.; Oue, N.; Sentani, K.; Sakamoto, N.; Uraoka, N.; Egi, H.; Hinoi, T.; Ohdan, H.; Yoshida, K.; Yasui, W. KIF11 is required for spheroid formation by oesophageal and colorectal cancer cells. Anticancer Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R.; Atefy, R.; Wagner, U.; Forster, T.; Fijan, A.; Bruderer, J.; Wilber, K.; Mihatsch, M.J.; Gasser, T.; Sauter, G. HER-2 and TOP2A coamplification in urinary bladder cancer. Int. J. Cancer 2003. [Google Scholar] [CrossRef]
- Sønderstrup, I.M.H.; Nygård, S.B.; Poulsen, T.S.; Linnemann, D.; Stenvang, J.; Nielsen, H.J.; Bartek, J.; Brünner, N.; Nørgaard, P.; Riis, L. Topoisomerase-1 and -2A gene copy numbers are elevated in mismatch repair-proficient colorectal cancers. Mol. Oncol. 2015. [Google Scholar] [CrossRef]
- Bofin, A.M.; Ytterhus, B.; Hagmar, B.M. TOP2A and HER-2 gene amplification in fine needle aspirates from breast carcinomas. Cytopathology 2003. [Google Scholar] [CrossRef] [PubMed]
- Coss, A.; Tosetto, M.; Fox, E.J.; Sapetto-Rebow, B.; Gorman, S.; Kennedy, B.N.; Lloyd, A.T.; Hyland, J.M.; O’Donoghue, D.P.; Sheahan, K.; et al. Increased topoisomerase IIα expression in colorectal cancer is associated with advanced disease and chemotherapeutic resistance via inhibition of apoptosis. Cancer Lett. 2009. [Google Scholar] [CrossRef]
- Tao, J.; Zhi, X.; Tian, Y.; Li, Z.; Zhu, Y.; Wang, W.; Xie, K.; Tang, J.; Zhang, X.; Wang, L.; et al. CEP55 contributes to human gastric carcinoma by regulating cell proliferation. Tumor Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, T.; Dai, X.; Xu, J. Lentivirus-mediated knockdown of CEP55 suppresses cell proliferation of breast cancer cells. Biosci. Trends 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slimane, S.N.; Marcel, V.; Fenouil, T.; Catez, F.; Saurin, J.C.; Bouvet, P.; Diaz, J.J.; Mertani, H.C. Ribosome Biogenesis Alterations in Colorectal Cancer. Cells 2020, 11, 2361. [Google Scholar] [CrossRef] [PubMed]
- Stedman, A.; Beck-Cormier, S.; Le Bouteiller, M.; Raveux, A.; Vandormael-Pournin, S.; Coqueran, S.; Lejour, V.; Jarzebowski, L.; Toledo, F.; Robine, S.; et al. Ribosome biogenesis dysfunction leads to p53-mediated apoptosis and goblet cell differentiation of mouse intestinal stem/progenitor cells. Cell Death Differ. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, M.L.; Mullany, L.E.; Wolff, R.K.; Sakoda, L.C.; Samowitz, W.S.; Herrick, J.S. The p53-signaling pathway and colorectal cancer: Interactions between downstream p53 target genes and miRNAs. Genomics 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Sun, X.Y.; Yang, S.M.; Wang, Q.; Wang, Z.Y. Peroxiredoxin 1 promoted tumor metastasis and angiogenesis in colorectal cancer. Pathol. Res. Pract. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Fu, Z.; Wang, H.; Feng, J.; Wei, J.; Guo, J. Peroxiredoxin 2 is upregulated in colorectal cancer and contributes to colorectal cancer cells’ survival by protecting cells from oxidative stress. Mol. Cell. Biochem. 2014. [Google Scholar] [CrossRef]
- Verset, L.; Tommelein, J.; Moles Lopez, X.; Decaestecker, C.; Mareel, M.; Bracke, M.; Salmon, I.; De Wever, O.; Demetter, P. Epithelial expression of FHL2 is negatively associated with metastasis-free and overall survival in colorectal cancer. Br. J. Cancer 2013. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, Y.; Wang, X.; Yang, Q. CDK1 and CDC20 overexpression in patients with colorectal cancer are associated with poor prognosis: Evidence from integrated bioinformatics analysis. World J. Surg. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Xie, Y.J.; Zhang, X.K.; Huang, T.J.; Xu, H.F.; Mei, Y.; Liang, H.; Hu, H.; Lin, S.T.; Luo, F.F.; et al. GTSE1 is involved in breast cancer progression in p53 mutation-dependent manner. J. Exp. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Takeda, K.; Oda, E.; Tanaka, H.; Murasawa, H.; Takaoka, A.; Morishita, Y.; Akira, S.; Taniguchi, T.; Tanaka, N. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Dataset | # Samples (Cases/Control) | # Genes | Platform | Source | DEGs (UR/DR) |
|---|---|---|---|---|---|---|
| Group-1 (CA) | GSE8671 | 64 (32/32) | 54675 | GPL570 | [15] | 5498 (1355/4143) |
| GSE20916 | 69 (45/24) | 27697 | GPL570 | [16] | 2761 (1290/1471) | |
| GSE89076 | 80 (41/39) | 58717 | GPL16699 | [17] | 7538 (2844/4694) | |
| Group-2 (CAC) | GSE20842 | 130 (65/65) | 40645 | GPL4133 | [18] | 3112 (1432/1680) |
| GSE20916 | 60 (36/24) | 27697 | GPL570 | [16] | 3556 (1820/1736) | |
| GSE39582 | 585 (566/19) | 54675 | GPL570 | [19] | 3389 (1582/1807) | |
| GSE110225 | 34 (17/17) | 54675 | GPL570 | [20] | 1109 (478/631) | |
| TCGA-COAD | 519 (478/41) | 56499 | Illumina | [21] | 6703 (3180/3523) | |
| TCGA-READ | 176 (166/10) | 56493 | Illumina | [22] | 6813 (3291/3522) | |
| Group-3 (CC) | GSE3964 | 30 (18/12) | 23232 | GPL3282 | [23] | 483 (158/325) |
| GSE113513 | 28 (14/14) | 49395 | GPL15207 | Unpublished | 2864 (1151/1713) | |
| GSE32323 | 44 (17/27) | 54675 | GPL570 | [24] | 4671 (4530/141) | |
| GSE21510 | 148 (123/25) | 54675 | GPL570 | [25] | 7720 (4383/3137) |
| Oncomine Groups | Dataset | # Samples (Cases/Control) | Oxidative Stress DEGs (UR/DR) | Apoptosis DEGs (UR/DR) |
|---|---|---|---|---|
| Oncomine Group-1 (CA) | Sabates (GSE8671) | 64 (32/32) | 29 (21/08) | 208 (142/66) |
| Skrzypczak (GSE20916) | 69 (45/24) | 24 (15/09) | 189 (105/84) | |
| Skrzypczak2 (GSE20916) | 15 (5/10) | 22 (12/10) | 176 (90/86) | |
| Oncomine Group-2 (CAC) | Dulak (GSE36458) | 122 (62/60) | 19 (10/09) | 175 (93/82) |
| Gaedcke (GSE20842) | 130 (65/65) | 39 (23/16) | 199 (107/92) | |
| Kaiser (GSE5206) | 54 (49/5) | 37 (20/17) | 225 (117/108) | |
| Kurashina (GSE11417) | 184 (94/90) | 29 (18/11) | 173 (111/62) | |
| Skrzypczak (GSE20916) | 60 (36/24) | 27 (15/12) | 187 (101/86) | |
| TCGA CRC | 184 (162/22) | 38 (21/17) | 257 (120/137) | |
| TCGA CRC2 | 970 (389/581) | 33 (17/16) | 209 (108/101) | |
| Oncomine Group-3 (CC) | Graudens (GSE3964) | 30 (18/12) | 16 (09/07) | 64 (20/44) |
| Hong (GSE9348) | 82 (70/12) | 28 (16/12) | 180 (110/70) | |
| Skrzypczak2 (GSE20916) | 15 (5/10) | 30 (15/15) | 188 (97/91) |
| Groups | Robust UR | Robust DR | Total Robust DEGs |
|---|---|---|---|
| G1 (CA) | 186 | 449 | 635 |
| G2 (CAC) | 499 | 494 | 993 |
| G3 (CC) | 206 | 79 | 285 |
| Group | Term | KEGG Pathway | Count | p-Value | Genes |
|---|---|---|---|---|---|
| G1 | hsa03008 | Ribosome biogenesis in eukaryotes | 7 | 2.92 × 10−7 | RBM28, NOB1, HEATR1, NHP2, WDR75, UTP14A, RAN |
| hsa04062 | Chemokine signaling pathway | 7 | 2.48 × 10−5 | CXCL12, ADCY9, GNG2, CCL21, GNG7, CXCL3, CCR2 | |
| hsa05200 | Pathways in cancer | 8 | 0.000208 | CXCL12, ADCY9, GNG2, MYC, GNG7, LPAR1, IGF1, FOXO1 | |
| hsa04151 | PI3K-Akt signaling pathway | 5 | 0.025838 | GNG2, MYC, GNG7, LPAR1, IGF1 | |
| hsa04727 | GABAergic synapse | 3 | 0.032272 | ADCY9, GNG2, GNG7 | |
| hsa05032 | Morphine addiction | 3 | 0.036574 | ADCY9, GNG2, GNG7 | |
| hsa04713 | Circadian entrainment | 3 | 0.03956 | ADCY9, GNG2, GNG7 | |
| hsa04723 | Retrograde endocannabinoid signaling | 3 | 0.044207 | ADCY9, GNG2, GNG7 | |
| hsa04060 | Cytokine-cytokine receptor interaction | 4 | 0.045786 | CXCL12, CCL21, CXCL3, CCR2 | |
| G2 | hsa04110 | Cell cycle | 14 | 2.75 × 10−19 | PLK1, TTK, CDC6, CCNA2, CDC20, CCNB1, CDC45, PTTG1, CHEK1, CDK1, MCM4, BUB1, MCM2, MAD2L1 |
| hsa04114 | Oocyte meiosis | 8 | 1.01 × 10−8 | CDC20, CCNB1, PTTG1, PLK1, CDK1, BUB1, MAD2L1, AURKA | |
| hsa04914 | Progesterone-mediated oocyte maturation | 6 | 2.91 × 10−6 | CCNA2, CCNB1, PLK1, CDK1, BUB1, MAD2L1 | |
| hsa04115 | p53 signaling pathway | 4 | 0.000765 | CCNB1, RRM2, CHEK1, CDK1 | |
| hsa05203 | Viral carcinogenesis | 4 | 0.01777 | CCNA2, CDC20, CHEK1, CDK1 | |
| hsa05166 | HTLV-I infection | 4 | 0.031122 | CDC20, PTTG1, CHEK1, MAD2L1 | |
| G3 | hsa03008 | Ribosome biogenesis in eukaryotes | 9 | 1.05 × 10−11 | RCL1, NOP58, WDR3, HEATR1, RPP40, UTP14A, GTPBP4, WDR43, RAN |
| hsa00240 | Pyrimidine metabolism | 4 | 0.001799 | RRM2, POLR1B, POLR1C, POLR1D | |
| hsa03020 | RNA polymerase | 3 | 0.00273 | POLR1B, POLR1C, POLR1D | |
| hsa00230 | Purine metabolism | 4 | 0.008596 | RRM2, POLR1B, POLR1C, POLR1D |
| Groups | Robust Oxidative stress DEGs (UR/DR) | Robust Apoptosis DEGs (UR/DR) |
|---|---|---|
| G1 | 04 (02/02) | 28 (19/09) |
| G2 | 09 (04/05) | 47 (23/24) |
| G3 | 02 (00/02) | 18 (09/09) |
| Groups | Robust Oxidative-Stress DEGs | p-Value | Fold Change |
|---|---|---|---|
| G1 | GPX2 | 4.95 × 10−6 | 2.282463 |
| KIF9 | 3.95 × 10−6 | 2.274907 | |
| PRDX6 | 1.23 × 10−7 | −2.7178 | |
| SEPP1 | 0.000103 | −4.44091 | |
| G2 | FOXM1 | 0.009917 | 1.598071 |
| GSS | 0.00024 | 1.346084 | |
| NUDT1 | 0.000948 | 1.576259 | |
| PRDX2 | 0.000454 | 1.372055 | |
| ANGPTL7 | 0.005649 | −4.24904 | |
| MSRA | 7.11 × 10−5 | −1.59741 | |
| PDLIM1 | 0.002234 | −1.33504 | |
| PRDX6 | 0.00074 | −2.2563 | |
| SCARA3 | 0.000324 | −1.59527 | |
| G3 | CCL5 | 2.39 × 10−5 | −1.84543 |
| SEPP1 | 8.97 × 10−7 | −4.10094 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, A.R.; Leung, M.-Y.; Roy, S. Identification of Hub Genes in Different Stages of Colorectal Cancer through an Integrated Bioinformatics Approach. Int. J. Environ. Res. Public Health 2021, 18, 5564. https://doi.org/10.3390/ijerph18115564
Patil AR, Leung M-Y, Roy S. Identification of Hub Genes in Different Stages of Colorectal Cancer through an Integrated Bioinformatics Approach. International Journal of Environmental Research and Public Health. 2021; 18(11):5564. https://doi.org/10.3390/ijerph18115564
Chicago/Turabian StylePatil, Abhijeet R., Ming-Ying Leung, and Sourav Roy. 2021. "Identification of Hub Genes in Different Stages of Colorectal Cancer through an Integrated Bioinformatics Approach" International Journal of Environmental Research and Public Health 18, no. 11: 5564. https://doi.org/10.3390/ijerph18115564
APA StylePatil, A. R., Leung, M. -Y., & Roy, S. (2021). Identification of Hub Genes in Different Stages of Colorectal Cancer through an Integrated Bioinformatics Approach. International Journal of Environmental Research and Public Health, 18(11), 5564. https://doi.org/10.3390/ijerph18115564