
Co-Circulation of Bovine Leukemia Virus Haplotypes among Humans, Animals, and Food Products: New Insights of Its Zoonotic Potential

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population and Samples
2.2. Data Retrieval and Viral Target Region
2.3. Phylogenetic Analyses
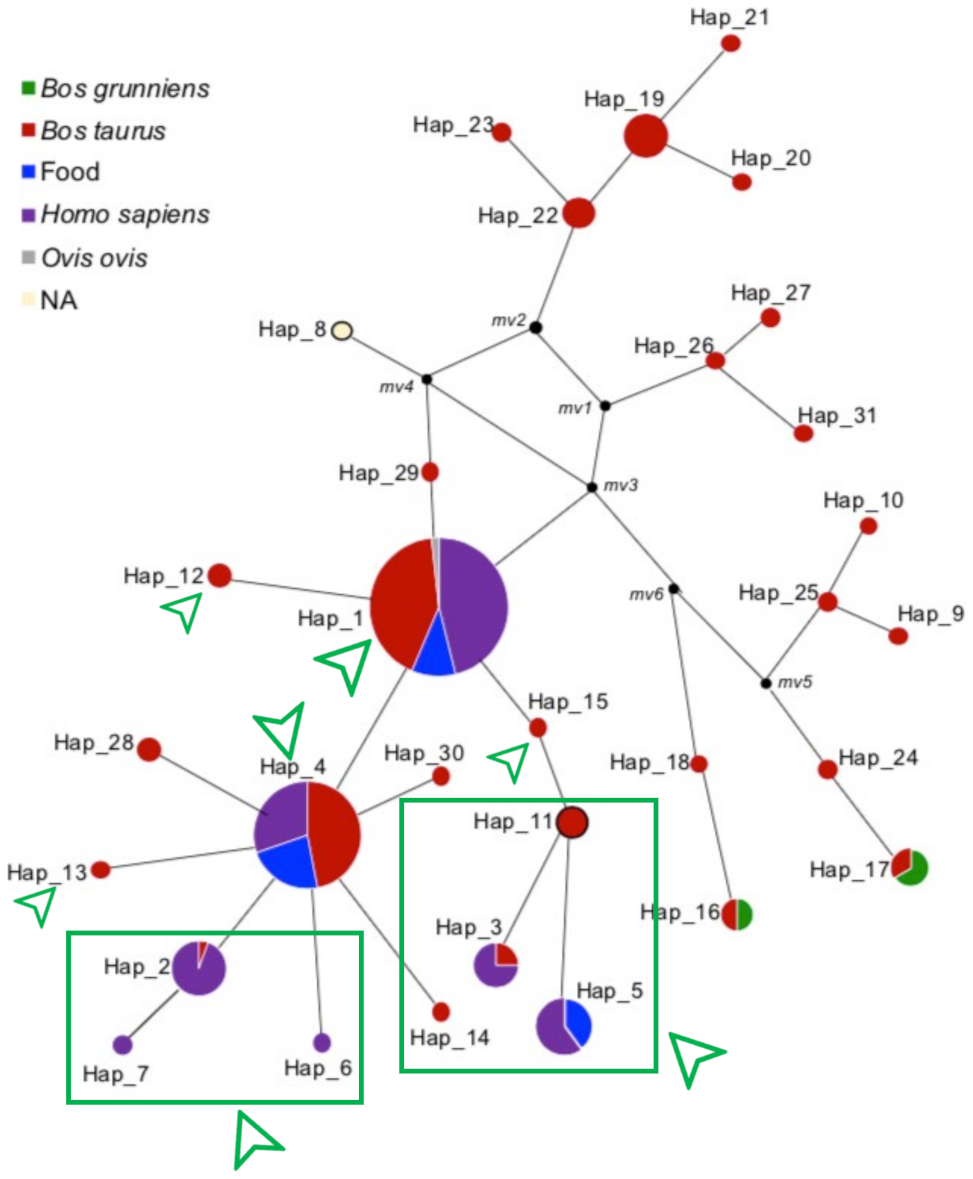
2.4. Haplotype Distribution and Network Analysis
2.5. Identification of Recombination Events between Colombian Isolates Obtained from Cattle, Food Products and Humans
2.6. Identification of Risks of Exposure to BLV in Humans and Association with Circulating Haplotypes
3. Results
3.1. Phylogenetic Analyses
3.2. Haplotype Distribution and Network Analysis
3.3. Identification of Recombination Events
3.4. Risks of Exposure to BLV and Haplotypes Association
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization Zoonoses; World Health Organization: Geneva, Switzerland, 2017.
- CDC. Zoonotic Diseases, One Health; CDC: Atlanta, GA, USA. Available online: https://www.cdc.gov/onehealth/basics/zoonotic-diseases.html (accessed on 14 January 2020).
- Teshome, H. Review on Principles of Zoonoses Prevention, Control and Eradication. Am. J. Biomed. Sci. Res. 2019, 3, 188–197. [Google Scholar] [CrossRef]
- Bauerfeind, R.; von Graevenitz, A.; Kimmig, P.; Gerd-Schiefer, H.; Schwarz, T.; Slenczka, W.; Zahner, H. Zoonoses: Infectious Diseases Transmissible from Animals to Humans, 4th ed.; Bauerfeind, R., von Graevenitz, A., Kimmig, P., Gerd Schiefer, H., Schwarz, T., Slenczka, W., Zahner, H., Eds.; ASM Press: Washington, DC, USA, 2016; ISBN 9781555819255. [Google Scholar]
- Karesh, W.B.; Dobson, A.; Lloyd-Smith, J.O.; Lubroth, J.; Dixon, M.A.; Bennett, M.; Aldrich, S.; Harrington, T.; Formenty, P.; Loh, E.H.; et al. Ecology of zoonoses: Natural and unnatural histories. Lancet 2012, 380, 1936–1945. [Google Scholar] [CrossRef]
- Wang, L.-F.; Crameri, G. Emerging zoonotic viral diseases. Rev. Sci. Technol. Off. Int. Epiz. 2014, 33, 569–581. [Google Scholar] [CrossRef]
- Barez, P.-Y.; de Brogniez, A.; Carpentier, A.; Gazon, H.; Gillet, N.; Gutiérrez, G.; Hamaidia, M.; Jacques, J.-R.; Perike, S.; Neelature Sriramareddy, S.; et al. Recent Advances in BLV Research. Viruses 2015, 7, 6080–6088. [Google Scholar] [CrossRef] [PubMed]
- Polat, M.; Takeshima, S.; Aida, Y. Epidemiology and genetic diversity of bovine leukemia virus. Virol. J. 2017, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- LaDronka, R.M.; Ainsworth, S.; Wilkins, M.J.; Norby, B.; Byrem, T.M.; Bartlett, P.C. Prevalence of Bovine Leukemia Virus Antibodies in US Dairy Cattle. Vet. Med. Int. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Corredor-Figueroa, A.P.; Salas, S.; Olaya-Galán, N.N.; Quintero, J.S.; Fajardo, Á.; Soñora, M.; Moreno, P.; Cristina, J.; Sánchez, A.; Tobón, J.; et al. Prevalence and molecular epidemiology of bovine leukemia virus in Colombian cattle. Infect. Genet. Evol. 2020, 80, 104171. [Google Scholar] [CrossRef]
- Polat, M.; Takeshima, S.S.; Hosomichi, K.; Kim, J.; Miyasaka, T.; Yamada, K.; Arainga, M.; Murakami, T.; Matsumoto, Y.; Barra Diaz, V.; et al. A new genotype of bovine leukemia virus in South America identified by NGS-based whole genome sequencing and molecular evolutionary genetic analysis. Retrovirology 2016, 13, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Virol, A.; Heinecke, N.; Tórtora, J.; Martínez, H.A.; González, V.D.; Hugo, F.; González-Fernández, V.D.; Ramírez, H. Detection and genotyping of bovine leukemia virus in Mexican cattle. Arch. Virol. 2017, 162, 3191–3196. [Google Scholar] [CrossRef]
- Selim, A.; Marawan, M.A.; Ali, A.F.; Manaa, E.; AbouelGhaut, H.A. Seroprevalence of bovine leukemia virus in cattle, buffalo, and camel in Egypt. Trop. Anim. Health Prod. 2020, 52, 1207–1210. [Google Scholar] [CrossRef]
- Feliziani, F.; Martucciello, A.; Iscaro, C.; Vecchio, D.; Petrini, S.; Grassi, C.; Bazzucchi, M.; De Carlo, E. Bovine leukemia virus: Experimental infection in buffaloes and evaluation of diagnostic test reliability. Res. Vet. Sci. 2017, 114, 450–454. [Google Scholar] [CrossRef]
- Lee, L.C.; Scarratt, W.K.; Buehring, G.C.; Saunders, G.K. Bovine leukemia virus infection in a juvenile alpaca with multicentric lymphoma. Can. Vet. J. Rev. Vét. Can. 2012, 53, 283–286. [Google Scholar]
- Olson, C.; Kettmann, R.; Burny, A.; Kaja, R. Goat lymphosarcoma from bovine leukemia virus. J. Natl. Cancer Inst. 1981, 67, 671–675. [Google Scholar]
- Nekoei, S.; Hafshejani, T.T.; Doosti, A.; Khamesipour, F. Molecular detection of Bovine leukemia virus in peripheral blood of Iranian cattle, camel and sheep. Pol. J. Vet. Sci. 2015, 18, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammerickx, M.; Portetelle, D.; Burny, A. Experimental Cross-Transmissions of Bovine Leukemia Virus (BLV) between Several Animal Species. Zent. Vet. R. B 1981, 28, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ikeda, H.; Mase, M. Restricted viral cDNA synthesis in cell lines that fail to support productive infection by bovine leukemia virus. Arch. Virol. 2018, 163, 2415–2422. [Google Scholar] [CrossRef] [PubMed]
- Reichert, M. Proteome analysis of sheep B lymphocytes in the course of bovine leukemia virus-induced leukemia. Exp. Biol. Med. 2017, 242, 1363–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corredor, A.P.; Gonzales, J.; Baquero, L.A.; Curtidor, H.; Olaya-Galán, N.N.; Patarroyo, M.A.; Gutierrez, M.F.; González, J.; Baquero, L.A.; Curtidor, H.; et al. In silico and in vitro analysis of boAP3d1 protein interaction with bovine leukaemia virus gp51. PLoS ONE 2018, 13, e0199397. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Sato, H.; Kubo, Y.; Wada, S.; Aida, Y. CAT1/SLC7A1 acts as a cellular receptor for bovine leukemia virus infection. FASEB J. 2019, fj201901528R. [Google Scholar] [CrossRef] [Green Version]
- Delarmelina, E.; Buzelin, M.A.; de Souza, B.S.; Souto, F.M.; Bicalho, J.M.; Falcão Câmara, R.J.; Resende, C.F.; Bueno, B.L.; Victor, R.M.; Florentino Galinari, G.C.; et al. High positivity values for bovine leukemia virus in human breast cancer cases from Minas Gerais, Brazil. PLoS ONE 2020, 15, e0239745. [Google Scholar] [CrossRef] [PubMed]
- Mesa, G.; Ulloa, J.C.; Uribe, A.M.; Gutierrez, M.F.; Giovanna, M.; Carlos, U.J.; María, U.A.; Gutierrez, M.F. Bovine Leukemia Virus Gene Segment Detected in Human Breast Tissue. Open J. Med. Microbiol. 2013, 3, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Khalilian, M.; Hosseini, S.M.; Madadgar, O. Bovine leukemia virus detected in the breast tissue and blood of Iranian women. Microb. Pathog. 2019, 135, 103566. [Google Scholar] [CrossRef]
- Lendez, P.A.; Martinez-Cuesta, L.; Nieto Farias, M.V.; Shen, H.; Dolcini, G.L.; Buehring, G.C.; Ceriani, M.C. Bovine leukemia virus presence in breast tissue of Argentinian women. Its association with cell proliferation and prognosis markers. Multidiscip. Cancer Investig. 2018, 2, 16–24. [Google Scholar] [CrossRef]
- Buehring, G.C.; Shen, H.M.; Jensen, H.M.; Choi, K.Y.; Sun, D.; Nuovo, G. Bovine Leukemia Virus DNA in Human Breast Tissue. Emerg. Infect. Dis. 2014, 20, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buehring, G.C.; Philpott, S.M.; Choi, K.Y. Humans have antibodies reactive with Bovine leukemia virus. AIDS Res. Hum. Retrovir. 2003, 19, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Buehring, G.C.; Sans, H.M. Breast cancer gone viral? Review of possible role of bovine leukemia virus in breast cancer, and related opportunities for cancer prevention. Int. J. Environ. Res. Public Health 2020, 17, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatami, A.; Pormohammad, A.; Farzi, R.; Saadati, H.; Mehrabi, M.; Kiani, S.J.; Ghorbani, S. Bovine Leukemia virus (BLV) and risk of breast cancer: A systematic review and meta-analysis of case-control studies. Infect. Agents Cancer 2020, 15, 1–8. [Google Scholar] [CrossRef]
- Robinson, L.A.; Jaing, C.J.; Pierce Campbell, C.; Magliocco, A.; Xiong, Y.; Magliocco, G.; Thissen, J.B.; Antonia, S. Molecular evidence of viral DNA in non-small cell lung cancer and non-neoplastic lung. Br. J. Cancer 2016, 115, 497–504. [Google Scholar] [CrossRef]
- Kim, Y.; Pierce, C.M.; Robinson, L.A. Impact of viral presence in tumor on gene expression in non-small cell lung cancer. BMC Cancer 2018, 18, 843. [Google Scholar] [CrossRef]
- Olaya-Galán, N.N.; Salas-Cárdenas, S.P.; Corredor-Figueroa, A.P.; Rodriguez-Sarmiento, J.L.; Ibáñez-Pinilla, M.; Monroy, R.; Rubiano, W.; de la Peña, J.; Shen, H.; Buehring, G.C.; et al. Evidence of bovine leukaemia virus in blood and breast tissues in Colombian women, a risk factor associated with breast cancer. J. Cancer Res. Clin. Oncol. 2021. submitted. [Google Scholar]
- Olaya-Galán, N.N.; Corredor-Figueroa, A.P.; Guzmán-Garzón, T.C.; Ríos-Hernandez, K.S.; Salas-Cárdenas, S.P.; Patarroyo, M.A.; Gutierrez, M.F. Bovine leukaemia virus DNA in fresh milk and raw beef for human consumption. Epidemiol. Infect. 2017, 145, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef]
- Bandelt, H.-J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Fisher, C.R.; Streicker, D.G.; Schnell, M.J. The spread and evolution of rabies virus: Conquering new frontiers. Nat. Rev. Microbiol. 2018, 16, 241–255. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 2020, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Szabo, K.; Trojnar, E.; Anheyer-Behmenburg, H.; Binder, A.; Schotte, U.; Ellerbroek, L.; Klein, G.; Johne, R. Detection of hepatitis E virus RNA in raw sausages and liver sausages from retail in Germany using an optimized method. Int. J. Food Microbiol. 2015, 215, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolo, I.; Angeloni, G.; Ponterio, E.; Ostanello, F.; Ruggeri, F.M. Detection of hepatitis E virus in pork liver sausages. Int. J. Food Microbiol. 2015, 193, 29–33. [Google Scholar] [CrossRef]
- Plowright, R.K.; Parrish, C.R.; McCallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-Smith, J.O. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degeling, C.; Johnson, J.; Kerridge, I.; Wilson, A.; Ward, M.; Stewart, C.; Gilbert, G. Implementing a One Health approach to emerging infectious disease: Reflections on the socio-political, ethical and legal dimensions. BMC Public Health 2015, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Viral Presence | |||
|---|---|---|---|
| Positive | Negative | p Value | |
| n (%) | n (%) | ||
| Pathology diagnoses | |||
| Malignant samples (n = 75) | 46 (61.3) | 29 (38.7) | <0.001 |
| Benign samples (n = 85) | 41 (48.8) | 44 (51.2) | |
| Age | |||
| ≥50 | 39 (63.9) | 22 (36.1) | 0.039 * |
| <50 | 61 (49.6) | 62 (50.4) | |
| Socio-demographic characteristics | |||
| Origin | 0.036 * | ||
| Bogotá | 61 (49.3) | 62 (50.4) | |
| Other | 30 (66.7) | 15 (33.3) | |
| Educational level | 0.785 | ||
| Elementary school | 23 (52.3) | 21 (47.7) | |
| High School | 34 (58.6) | 24 (41.4) | |
| Vocational and professional studies | 34 (53.1) | 30 (46.9) | |
| Risks of exposure to BLV | |||
| Dairy Products consumption | |||
| Flavored Yoghurt | 70 (59.8) | 47 (40.2) | 0.023 * |
| Home-made natural yoghurt (Kumis) | 61 (60.4) | 40 (39.6) | 0.042 * |
| Cheese | 83 (55.0) | 68 (45.0) | 0.614 |
| Jelly foot dessert (Gelatina de pata) | 41 (60.3) | 27 (39.7) | 0.148 |
| Industrialized milk | 90 (54.2) | 76 (45.8) | 0.708 |
| Direct contact with cattle | 39 (55.7) | 31 (44.3) | 0.428 |
| Amount of dairy products and raw milk | 0.04 * | ||
| 4 or more | 31 (72.1) | 12 (27.9) | |
| 3 | 18 (52.9) | 16 (47.1) | |
| 2 | 4 (30.8) | 9 (69.2) | |
| None | 2 (66.7) | 1 (33.3) | |
| Variables | Viral Presence | ||
|---|---|---|---|
| β | OR (95% CI) | p Value | |
| Age | |||
| ≥50 | 0.794 | 2.212 (1.111-4.402) | 0.024 |
| <50 | -- | 1.00 (Reference) | -- |
| City of origin | |||
| Other | 0.800 | 2.224 (1.030-4.805) | 0.042 |
| Bogota | -- | 1.00 (Reference) | -- |
| Milk consumption and dairy products a | -- | -- | 0.037 ** |
| Only milk or 1 dairy product | −0.990 | 0.372 (0.103-1.346) | 0.132 |
| Two dairy products/milk | 0.000 | 1.00 (0.431-2.321) | 1.000 |
| Three or more dairy products/milk | 0.885 | 2.424 (1.063-5.527) ** | 0.035 ** |
| No consumption of dairy products | -- | 1.00 (Reference) | -- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corredor-Figueroa, A.P.; Olaya-Galán, N.N.; Velandia-Álvarez, S.; Muñoz, M.; Salas-Cárdenas, S.P.; Ibáñez-Pinilla, M.; Patarroyo, M.A.; Gutiérrez, M.F. Co-Circulation of Bovine Leukemia Virus Haplotypes among Humans, Animals, and Food Products: New Insights of Its Zoonotic Potential. Int. J. Environ. Res. Public Health 2021, 18, 4883. https://doi.org/10.3390/ijerph18094883
Corredor-Figueroa AP, Olaya-Galán NN, Velandia-Álvarez S, Muñoz M, Salas-Cárdenas SP, Ibáñez-Pinilla M, Patarroyo MA, Gutiérrez MF. Co-Circulation of Bovine Leukemia Virus Haplotypes among Humans, Animals, and Food Products: New Insights of Its Zoonotic Potential. International Journal of Environmental Research and Public Health. 2021; 18(9):4883. https://doi.org/10.3390/ijerph18094883
Chicago/Turabian StyleCorredor-Figueroa, Adriana P., Nury N. Olaya-Galán, Sebastian Velandia-Álvarez, Marina Muñoz, Sandra P. Salas-Cárdenas, Milcíades Ibáñez-Pinilla, Manuel A. Patarroyo, and Maria F. Gutiérrez. 2021. "Co-Circulation of Bovine Leukemia Virus Haplotypes among Humans, Animals, and Food Products: New Insights of Its Zoonotic Potential" International Journal of Environmental Research and Public Health 18, no. 9: 4883. https://doi.org/10.3390/ijerph18094883
APA StyleCorredor-Figueroa, A. P., Olaya-Galán, N. N., Velandia-Álvarez, S., Muñoz, M., Salas-Cárdenas, S. P., Ibáñez-Pinilla, M., Patarroyo, M. A., & Gutiérrez, M. F. (2021). Co-Circulation of Bovine Leukemia Virus Haplotypes among Humans, Animals, and Food Products: New Insights of Its Zoonotic Potential. International Journal of Environmental Research and Public Health, 18(9), 4883. https://doi.org/10.3390/ijerph18094883