
Electrochemical Method for Ease Determination of Sodium Diclofenac Trace Levels in Water Using Graphene—Multi-Walled Carbon Nanotubes Paste Electrode
.png)




Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Obtaining of Working CNT-Based Paste Electrodes
2.3. Structural and Morphological Characterization
2.4. Electrochemical Experiments
3. Results and Discussions
3.1. Morphological and Electrochemical Characterization
3.2. Development of DCF Electrochemical Detection
3.3. Preconcentration Step Prior to DPV Detection
- E1 = −0.400 V vs. Ag/AgCl for 0.10 s for renewing electrode surface;
- E2 = −0.020 V vs. Ag/AgCl for 0.05 s, representing the first step of DCF oxidation considered as the first detection potential;
- E3 = +0.600 V vs. Ag/AgCl for 0.05 s, considered the second detection potential due to second step of DCF oxidation;
- E4 = +1.500 V vs. Ag/AgCl for 0.10 s, applied to assure in situ electrode surface cleaning based on concomitant slight rate of oxygen evolution.
3.4. Testing Preconcentration Step-Based Multiple-Pulsed Amperometry in Tap Water
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nannou, C.I.; Kosma, C.I.; Albanis, T.A. Occurrence of pharmaceuticals in surface waters: Analytical method development and environmental risk assessment. Int. J. Environ. Anal. Chem. 2015, 95, 1242–1262. [Google Scholar] [CrossRef]
- Vasquez, M.I.; Lambrianides, A.; Schneider, M.; Kümmerer, K.; Fatta-Kassinos, D. Environmental side effects of pharmaceutical cocktails: What we know and what we should know. J. Hazard. Mater. 2014, 279, 169–189. [Google Scholar] [CrossRef] [PubMed]
- Musson, S.E.; Townsend, T.G. Pharmaceutical compound content of municipal solid waste. J. Hazard. Mater. 2009, 162, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Aus der Beek, T.; Weber, F.A.; Bergmann, A.; Hickmann, S.; Ebert, I.; Hein, A.; Küster, A. Pharmaceuticals in the environment-global occurrences and perspectives: Pharmaceuticals in the global environment. Environ. Toxicol. Chem. 2016, 35, 823–835. [Google Scholar] [CrossRef]
- Amos Sibeko, P.; Naicker, D.; Mdluli, P.S.; Madikizela, L.M. Naproxen, ibuprofen, and diclofenac residues in river water, sediments and Eichhornia crassipes of Mbokodweni river in South Africa: An initial screening. Environ. Forensics 2019, 20, 129–138. [Google Scholar] [CrossRef]
- Gouda, A.A.; Kotb El-Sayed, M.I.; Amin, A.S.; El Sheikh, R. Spectrophotometric and spectrofluorometric methods for the determination of non-steroidal anti-inflammatory drugs: A review. Arab. J. Chem. 2013, 6, 145–163. [Google Scholar] [CrossRef] [Green Version]
- Madikizela, L.M.; Chimuka, L. Determination of ibuprofen, naproxen and diclofenac in aqueous samples using a multi-template molecularly imprinted polymer as selective adsorbent for solid-phase extraction. J. Pharm. Biomed. 2016, 128, 210–215. [Google Scholar] [CrossRef]
- Petrie, B.; Barden, R.; Kasprzyk-Hordern, B. A review on emerging contaminants in wastewaters and the environment: Current knowledge, understudied areas and recommendations for future monitoring. Water Res. 2015, 72, 3–27. [Google Scholar] [CrossRef] [PubMed]
- O’Flynn, D.; Lawler, J.; Yusuf, A.; Parle-McDermott, A.; Harold, D.; Mc Cloughlin, T.; Holland, L.; Regan, F.; White, B. A review of pharmaceutical occurrence and pathways in the aquatic environment in the context of a changing climate and the COVID-19 pandemic. Anal. Methods 2021, 13, 575–594. [Google Scholar] [CrossRef]
- Madikizela, L.M.; Chimuka, L. Simultaneous determination of naproxen, ibuprofen and diclofenac in wastewater using solid-phase extraction with high performance liquid chromatography. Water SA 2017, 43, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Samaras, V.G.; Thomaidis, N.S.; Stasinakis, A.S.; Gatidou, G.; Lekkas, T.D. Determination of selected non-steroidal anti-inflammatory drugs in wastewater by gas chromatography-mass spectrometry. Int. J. Environ. Anal. Chem. 2010, 90, 219–229. [Google Scholar] [CrossRef]
- Maurer, H.H.; Tauvel, F.X.; Kraemer, T. Screening procedure for detection of non-steroidal anti-inflammatory drugs and their metabolites in urine as part of a systematic toxicological analysis procedure for acidic drugs and poisons by gas chromatography- mass spectrometry after extractive methylation. J. Anal. Toxicol. 2001, 25, 237–244. [Google Scholar] [PubMed] [Green Version]
- Wolecki, D.; Caban, M.; Pazdro, K.; Mulkiewicz, E.; Stepnowski, P.; Kumirska, J. Simultaneous determination of non-steroidal anti-inflammatory drugs and natural estrogens in the mussels Mytilus edulis trossulus. Talanta 2019, 200, 316–323. [Google Scholar] [CrossRef]
- Patel, P.N.; Samanthula, G.; Shrigod, V.; Modh, S.C.; Chaudhari, J.R. RP-HPLC method for determination of several NSAIDs and their combination drugs. Chromatogr. Res. Int. 2013, 2013, 242868. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Myung, S.-W. Simultaneous determination of nonsteroidal anti-inflammatory drugs in aqueous samples using dispersive liquid-liquid microextraction and HPLC analysis. Bull. Korean Chem. Soc. B 2015, 36, 2901–2906. [Google Scholar] [CrossRef]
- Quek, N.M.; Law, W.S.; Lau, H.F.; Zhao, J.H.; Hauser, P.C.; Li, S.F.Y. Determination of pharmaceuticals classified as emerging pollutants using capillary electrophoresis with capacitively coupled contactless conductivity detection. Electrophoresis 2008, 29, 3701–3709. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.A. Nanomaterials for pharmaceuticals determination. Bioenergetics 2016, 5, 1000226. [Google Scholar] [CrossRef]
- Kurbanoglu, S.; Ozkan, S.A. Electrochemical carbon based nanosensors: A promising tool in pharmaceutical and biomedical analysis. J. Pharm. Biomed. Anal. 2018, 147, 439–457. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Z. Application of electrochemical sensors based on carbon nanomaterials for detection of flavonoids. Nanomaterials 2020, 10, 2020. [Google Scholar] [CrossRef]
- Torrinha, A.; Oliveira, T.M.B.F.; Ribeiro, F.W.P.; Correia, A.N.; Lima-Neto, P.; Morais, S. Application of nanostructured carbon-based electrochemical (Bio)sensors for screening of emerging pharmaceutical pollutants in waters and aquatic species: A review. Nanomaterials 2020, 10, 1268. [Google Scholar] [CrossRef]
- Yang, X.; Feng, B.; He, X.; Li, F.; Ding, Y.; Fei, J. Carbon nanomaterial based electrochemical sensors for biogenic amines. Microchim. Acta 2013, 180, 935–956. [Google Scholar] [CrossRef]
- Power, A.C.; Gorey, B.; Chandra, S.; Chapman, J. Carbon nanomaterials and their application to electrochemical sensors: A review. Nanotechnol. Rev. 2018, 7, 19–41. [Google Scholar] [CrossRef]
- Xie, F.; Yang, M.; Jiang, M.; Huang, X.-J.; Liu, W.-Q.; Xie, P.-H. Carbon based nanomaterials—A promising electrochemical sensor toward persistent toxic substance. Trends Analyt. Chem. 2019, 119, 115624. [Google Scholar] [CrossRef]
- Siqueira, J.R., Jr.; de Oliveira, O.N., Jr. Nanostructures; Elsevier: Amsterdam, The Netherlands, 2017; pp. 233–249. [Google Scholar]
- Tiwari, J.N.; Vij, V.; Kemp, K.C.; Kim, K.S. Engineered carbon-nanomaterial-based electrochemical sensors for biomolecules. ACS Nano 2015, 10, 46–80. [Google Scholar] [CrossRef] [Green Version]
- Manea, F. Modern Electrochemical Methods in Nano, Surface and Corrosion Science; IntechOpen: Rijeka, Croatia, 2017; pp. 33–54. [Google Scholar]
- Pan, M.; Yin, Z.; Liu, K.; Du, X.; Liu, H.; Wang, S. Carbon-based nanomaterials in sensors for food safety. Nanomaterials 2019, 9, 1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motoc, S.; Manea, F.; Orha, C.; Pop, A. Enhanced electrochemical response of diclofenac at a fullerene-carbon nanofiber paste electrode. Sensors 2019, 19, 1332. [Google Scholar] [CrossRef] [Green Version]
- Arvand, M.; Gholizadeh, T.M.; Zanjanchi, M.A. MWCNTs/Cu(OH)2 nanoparticles/IL nanocomposite modified glassy carbon electrode as a voltammetric sensor for determination of the non-steroidal anti-inflammatory drug diclofenac. Mater. Sci. Eng. C 2012, 32, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Shalauddin, M.; Akhter, S.; Bagheri, S.; Karim, M.S.A.; Adib Kadri, N.; Basirun, W.J. Immobilized copper ions on MWCNTS-Chitosan thin film: Enhanced amperometric sensor for electrochemical determination of diclofenac sodium in aqueous solution. Int. J. Hydrogen Energy 2017, 42, 19951–19960. [Google Scholar] [CrossRef]
- Ensafi, A.A.; Izadi, M.; Karimi-Maleh, H. Sensitive voltammetric determination of diclofenac using room-temperature ionic liquid-modified carbon nanotubes paste electrode. Ionics 2012, 19, 137–144. [Google Scholar] [CrossRef]
- Eteya, M.M.; Rounaghi, G.H.; Deiminiat, B. Fabrication of a new electrochemical sensor based on Au Pt bimetallic nanoparticles decorated multi-walled carbon nanotubes for determination of diclofenac. Microchem. J. 2018, 144, 254. [Google Scholar] [CrossRef]
- Motoc, S.; Manea, F.; Iacob, A.; Martinez-Joaristi, A.; Gascon, J.; Pop, A.; Schoonman, J. Electrochemical selective and simultaneous detection of diclofenac and ibuprofen in aqueous solution using HKUST-1 metal-organic framework-carbon nanofiber composite elect. Sensors 2016, 16, 1719. [Google Scholar] [CrossRef] [Green Version]
- Ihos, M.; Remes, A.; Manea, F. Electrochemical determination of diclofenac using boron-doped diamond electrode. J. Environ. Prot. Ecol. 2012, 13, 2096. [Google Scholar]
- Manea, F.; Ihos, M.; Remes, A.; Burtica, G.; Schoonman, J. Electrochemical determination of diclofenac sodium in aqueous Solution on cu-doped zeolite-expanded graphite-epoxy electrode. Electroanalysis 2010, 22, 2058–2063. [Google Scholar] [CrossRef]
- Boumya, W.; Taoufik, N.; Achak, M.; Bessbousse, H.; Elhalil, A.; Barka, N. Electrochemical sensors and biosensors for the determination of diclofenac in pharmaceutical, biological and water samples. Talanta Open 2021, 3, 100026. [Google Scholar] [CrossRef]
- Voda, R.D.; Negrea, S.; Păcurariu, C.; Surdu, A.; Ianculescu, A.; Pop, A.; Manea, F. CuBi2O4 synthesis, characterization, and application in sensitive amperometric/voltammetric detection of amoxicillin in aqueous solutions. Nanomaterials 2021, 11, 740. [Google Scholar]
- Motoc, S.; Cretu, C.; Costisor, O.; Baciu, A.; Manea, F.; Szerb, E.I. Cu(I) coordination complex precursor for randomized CuOx microarray loaded on carbon nanofiber with excellent electrocatalytic performance for electrochemical glucose detection. Sensors 2019, 19, 5353. [Google Scholar] [CrossRef] [Green Version]
- Motoc, S.; Schinteie, B.; Pop, A.; Negrea, S.; Cretu, C.; Szerb, E.I.; Manea, F. Graphene quantum dots and Cu(I) liquid crystal for advanced electrochemical detection of doxorubicine in aqueous solutions. Nanomaterials 2021, 11, 2788. [Google Scholar] [CrossRef]
- Konopka, S.J.; McDuffie, B. Diffusion coefficients of ferri- and ferrocyanide ions in aqueous media, using twin-electrode thin-layer electrochemistry. Anal. Chem. 1970, 42, 1741–1746. [Google Scholar] [CrossRef]
- Negrea, S.; Diaconu, L.A.; Nicorescu, V.; Motoc, S.; Orha, C.; Manea, F. Graphene oxide electroreduced onto boron-doped diamond and electrodecorated with silver (Ag/GO/BDD) electrode for tetracycline detection in aqueous solution. Nanomaterials 2021, 11, 1566. [Google Scholar] [CrossRef]
- Mostofizadeh, A.; Li, Y.; Song, B.; Huang, Y. Synthesis, properties, and applications of low-dimensional carbon-related nanomaterials. J. Nanomater. 2011, 2011, 685081. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, J.-K.; Wu, Y.; Murray, R.W. Photoluminescence and electronic interaction of anthracene derivatives adsorbed on sidewalls of single-walled carbon nanotubes. Nano Lett. 2003, 3, 403–407. [Google Scholar] [CrossRef]
- Honakeri, N.C.; Malode, S.J.; Kulkarni, R.M.; Shetti, N.P. Electrochemical behavior of diclofenac sodium at coreshell nanostructure modified electrode and its analysis in human urine and pharmaceutical samples. Sens. Int. 2020, 1, 100002. [Google Scholar] [CrossRef]
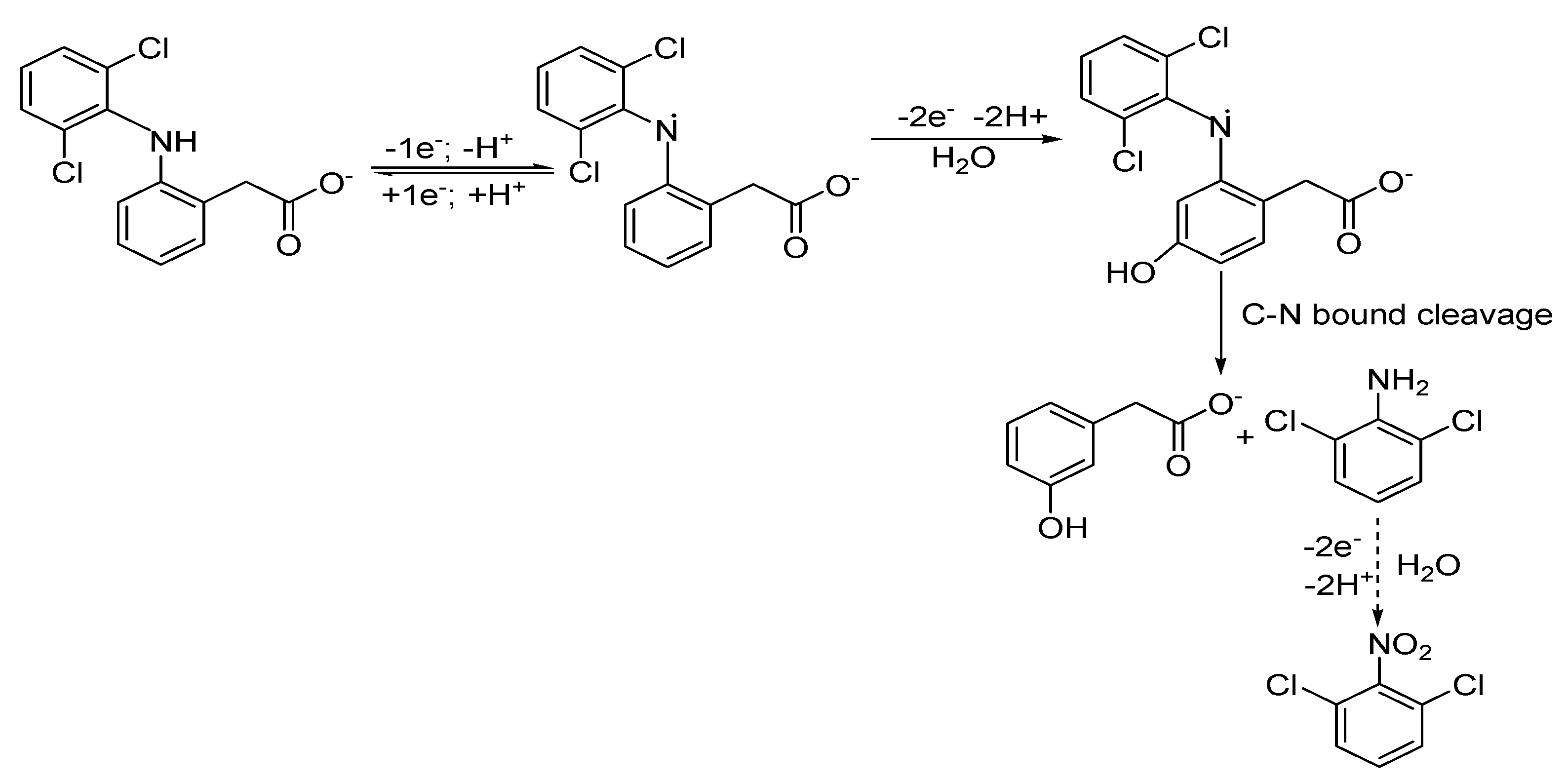
- Cid-Ceron, M.M.; Guzman-Hernzandez, D.S.; Ramirez-Silva, M.T.; Galano, A.; Romero-Romo, M.; Palomar-Pardave, M. New insigths on the kinetics and me-chanism of the electrochemical oxidation of diclofenac in neutral aqueous medium. Electrochim. Acta 2016, 199, 92–98. [Google Scholar] [CrossRef]
- Sanecki, P.; Skitał, P.; Kaczmarsk, K. Numerical modeling of ECE-ECE and parallel EE-EE mechanisms in cyclic voltammetry. reduction of 1,4-benzenedisulfonyl difluoride and 1,4-naphthalenedisulfonyl difluoride. Electroanalysis 2006, 18, 981–991. [Google Scholar] [CrossRef]
- Goyal, R.N.; Chatterjee, S.; Agrawal, B. Electrochemical investigations of diclofenac at edge plane pyrolytic graphite electrode and its determination in human urine. Sens. Actuators B Chem. 2010, 145, 743–748. [Google Scholar] [CrossRef]
- Aguilar-Lira, G.Y.; Alvarez-Romero, G.A.; Zamora-Suarez, A.; Palomar-Pardave, M.; Rojas-Hernández, A.; Rodriguez-Avila, J.A.; Paez-Hernandez, M.E. New insights on diclofenac electrochemistry using graphite as working electrode. J. Electroanal. Chem. 2017, 794, 182–188. [Google Scholar] [CrossRef]
- Sasal, A.; Tyszczuk-Rotko, K.; Wojciak, M.; Sowa, I. First electrochemical sensor (screen-printed carbon electrode modified with carboxyl functionalized multiwalled carbon nanotubes) for ultratrace determination of diclofenac. Materials 2020, 13, 781. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, A.; Karimi-Maleh, H.; Ensafi, A.A.; Beitollahi, H. Application of modified multiwall carbon nanotubes paste electrode for simultaneous voltammetric determination of morphine and diclofenac in biological and pharmaceutical samples. Sens. Actuators B Chem. 2012, 169, 96–105. [Google Scholar] [CrossRef]
- Okoth, O.K.; Yan, K.; Liu, L.; Zhang, J. Simultaneous electrochemical determination of paracetamol and diclofenac based on poly(diallyldimethylammonium chloride) functionalized graphene. Electroanalysis 2015, 28, 76–82. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight Ratio, % | |||
|---|---|---|---|
| Paste Electrode Type | Carbon Nanotubes (CNT) | Graphene (GR) | Paraffin Oil |
| CNT | 1 | - | 3 |
| GR-CNT | 1 | 1 | 3.5 |
| Electrode | Geometrical Surface Area/cm2 | Electroactive Surface Area/cm2 | Electroactive/Geometrical Surface Area Ratio |
|---|---|---|---|
| CNT | 0.0765 | 0.117 | 1.53 |
| GR-CNT | 0.0176 | 0.038 | 2.16 |
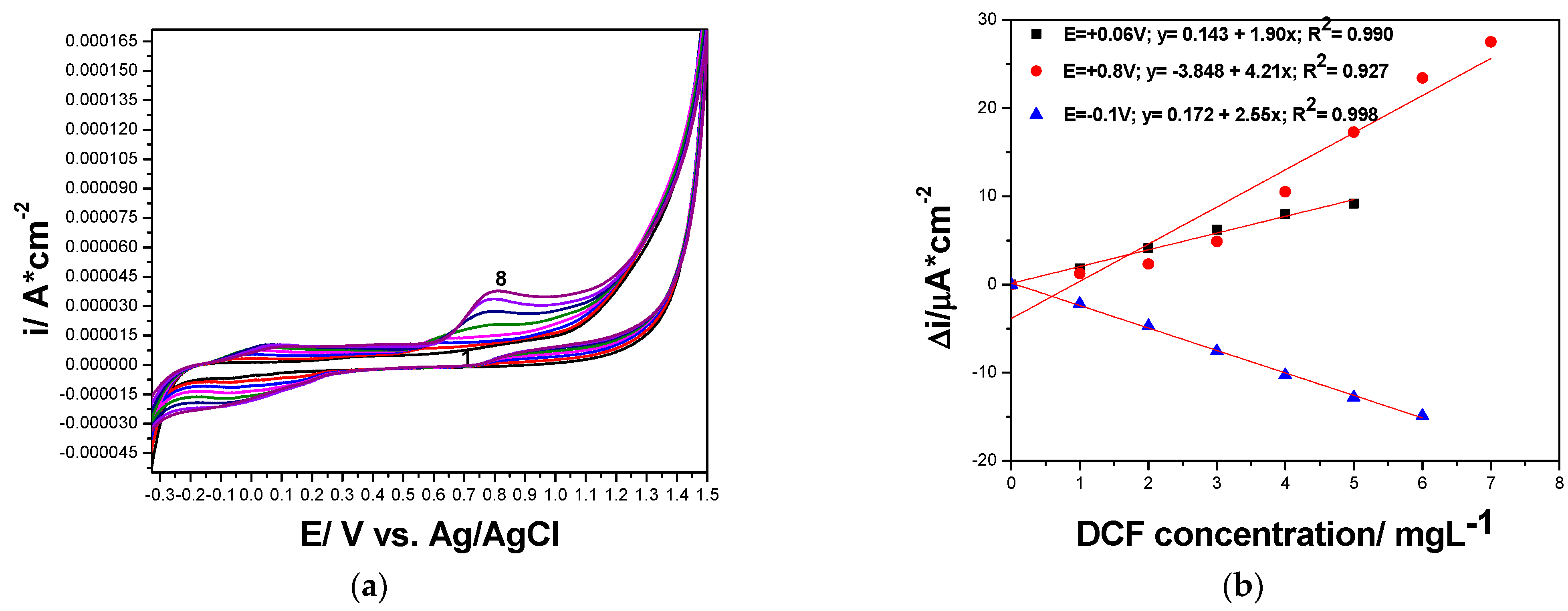
| Electrode Type | Potential Range | Edet (V vs. Ag/AgCl) | Sensitivity (µA/cm2·mg·L−1) |
|---|---|---|---|
| CNT | Anodic | +0.060 | 1.90 |
| Anodic | +0.800 | 4.21 | |
| Cathodic | −0.100 | 2.55 | |
| GR-CNT | Anodic | −0.050 | 10.3 |
| Anodic | +0.580 | 12.7 | |
| Anodic | +1.180 | 52.5 | |
| Cathodic | −0.200 | 10.7 |
| DCF Concentration, mg·L−1 | E = −0.050 V vs. Ag/AgCl | E = +0.580 V vs. Ag/AgCl | E = +1.150 V vs. Ag/AgCl | E = +1.350 V vs. Ag/AgCl |
|---|---|---|---|---|
| 0.500 | y1 = −0.934 + 4.17; R2 = 0.983 | y2 = −0.445 + 6.30×; R2 = 0.976 | y3 = −0.383 + 18.45×; R2 = 0.994 | - ** |
| 0.050 | y1 = 0.162 + 2.82×; R2 = 0.952 | y2= −0.292 + 0.081×; R2 = 0.967 | y3 = 0.778 + 21.55×; R2 = 0.995 | Y4 = 1.791 + 45.89×; R2 = 0.985 |
| 0.005 | - * | y2 = −0.191 + 2.76×; R2 = 0.992 | y2= −0.795 + 10.52×; R2 = 0.990 | - ** |
| Technique | Detection Potential/ V vs. Ag/AgCl | Sensitivity/ µA·mgL−1 | Correlation Coefficient/R2 | LOD [a]/ mg·L−1 | LQ [a]/ mg·L−1 | RSD [b] (%) |
|---|---|---|---|---|---|---|
| CV | −0.02 | 0.225 | 0.956 | 0.53 | 1.78 | 8.69 |
| −0.02 (C *) | 0.066 | 0.962 | 2.02 | 6.76 | 7.39 | |
| +0.53 | 0.378 | 0.982 | 0.43 | 1.44 | 9.14 | |
| +1.15 | 6.38 | 0.976 | 0.02 | 0.08 | 3.31 | |
| +1.35 | 9.40 | 0.998 | 0.01 | 0.05 | 1.31 | |
| DPV | +0.9 | 509.26 | 0.976 | 1.7 × 10−4 | 5.1 × 10−4 | 0.14 |
| Preconc. DPV | −0.21 | 31,400 | 0.954 | 1.4 × 10−6 | 4.6 × 10−6 | 0.22 |
| +0.9 | 51,000 | 0.946 | 2.3 × 10−6 | 7.8 × 10−6 | 0.35 | |
| SWV | +0.9 | 50.55 | 0.996 | 7.4 × 10−3 | 2.1 × 10−2 | 1.6 |
| MPA | +1.5 V | 13.15 | 0.983 | 2.8 × 10−2 | 9.4 × 10−2 | 0.18 |
| Preconc. MPA | +1.5 V | 347.32 | 0.968 | 5.9 × 10−4 | 1.97 × 10−3 | 0.48 |
| Method | Electrode | Modifier | LOD (ng·L−1) | Matrix | Ref. |
|---|---|---|---|---|---|
| DPAdSV | SPCE | COOH-CNT | 8.30 | River water | [49] |
| Preconc/SWV | CNF | Fullerene | 265 | Water | [14] |
| SWV | CPE | CNT-vinylferrocene | 590 × 103 | Tablets and urine | [50] |
| DPV | GCE | CNT/Cu(OH)2/1-EM1MPF6 | 11.80 × 103 | Ampoule, tablets, blood serum, fish serum, seawater | [29] |
| DPV | GCE | PDDA-Gr | 179.6 × 103 | Tablet, lake water | [51] |
| SWV | GCE | CNT-CHT | 6.20 × 103 | Tablets, urine | [30] |
| Prec. DPV | CNT | Graphene | 1.40 | Tap water | This work |
| Prec/MPA | CNT | Graphene | 590 | Tap water | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motoc, S.; Manea, F.; Baciu, A.; Orha, C.; Pop, A. Electrochemical Method for Ease Determination of Sodium Diclofenac Trace Levels in Water Using Graphene—Multi-Walled Carbon Nanotubes Paste Electrode. Int. J. Environ. Res. Public Health 2022, 19, 29. https://doi.org/10.3390/ijerph19010029
Motoc S, Manea F, Baciu A, Orha C, Pop A. Electrochemical Method for Ease Determination of Sodium Diclofenac Trace Levels in Water Using Graphene—Multi-Walled Carbon Nanotubes Paste Electrode. International Journal of Environmental Research and Public Health. 2022; 19(1):29. https://doi.org/10.3390/ijerph19010029
Chicago/Turabian StyleMotoc, Sorina, Florica Manea, Anamaria Baciu, Corina Orha, and Aniela Pop. 2022. "Electrochemical Method for Ease Determination of Sodium Diclofenac Trace Levels in Water Using Graphene—Multi-Walled Carbon Nanotubes Paste Electrode" International Journal of Environmental Research and Public Health 19, no. 1: 29. https://doi.org/10.3390/ijerph19010029
APA StyleMotoc, S., Manea, F., Baciu, A., Orha, C., & Pop, A. (2022). Electrochemical Method for Ease Determination of Sodium Diclofenac Trace Levels in Water Using Graphene—Multi-Walled Carbon Nanotubes Paste Electrode. International Journal of Environmental Research and Public Health, 19(1), 29. https://doi.org/10.3390/ijerph19010029