
The Potential Key Role of the NRF2/NQO1 Pathway in the Health Effects of Arsenic Pollution on SCC

 , ,
, , 
Abstract
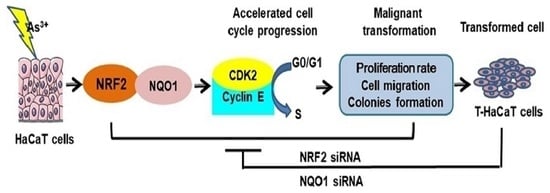
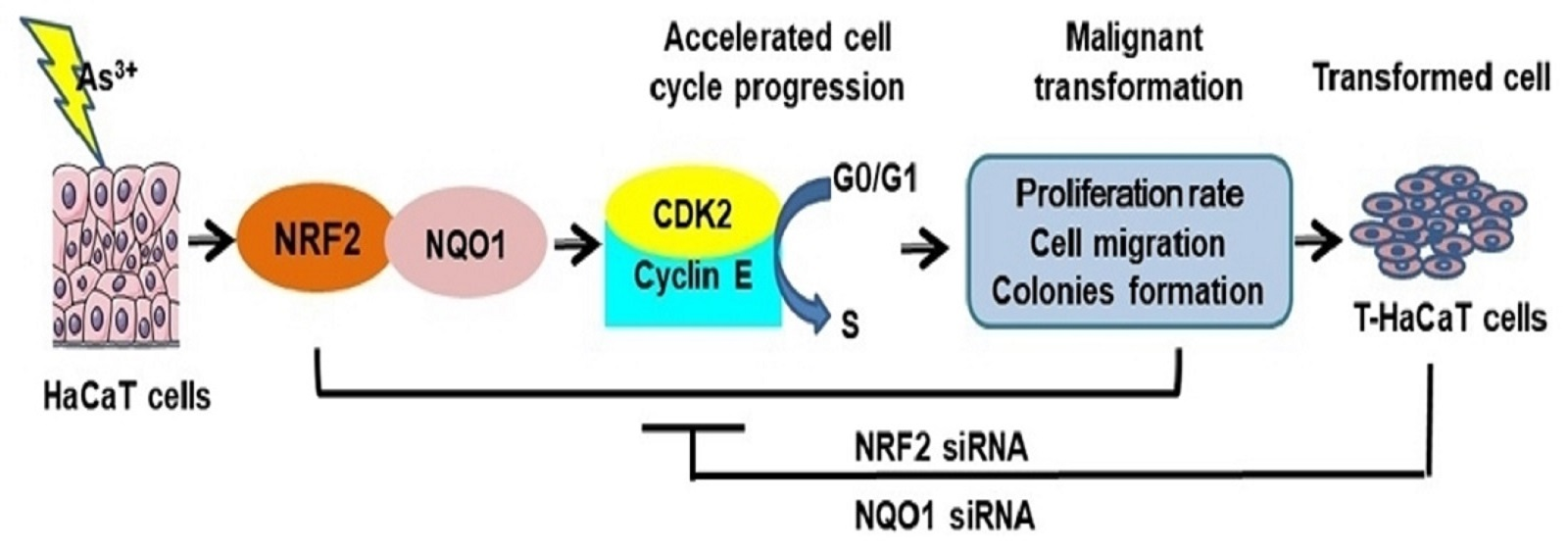
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Exposure to Arsenic
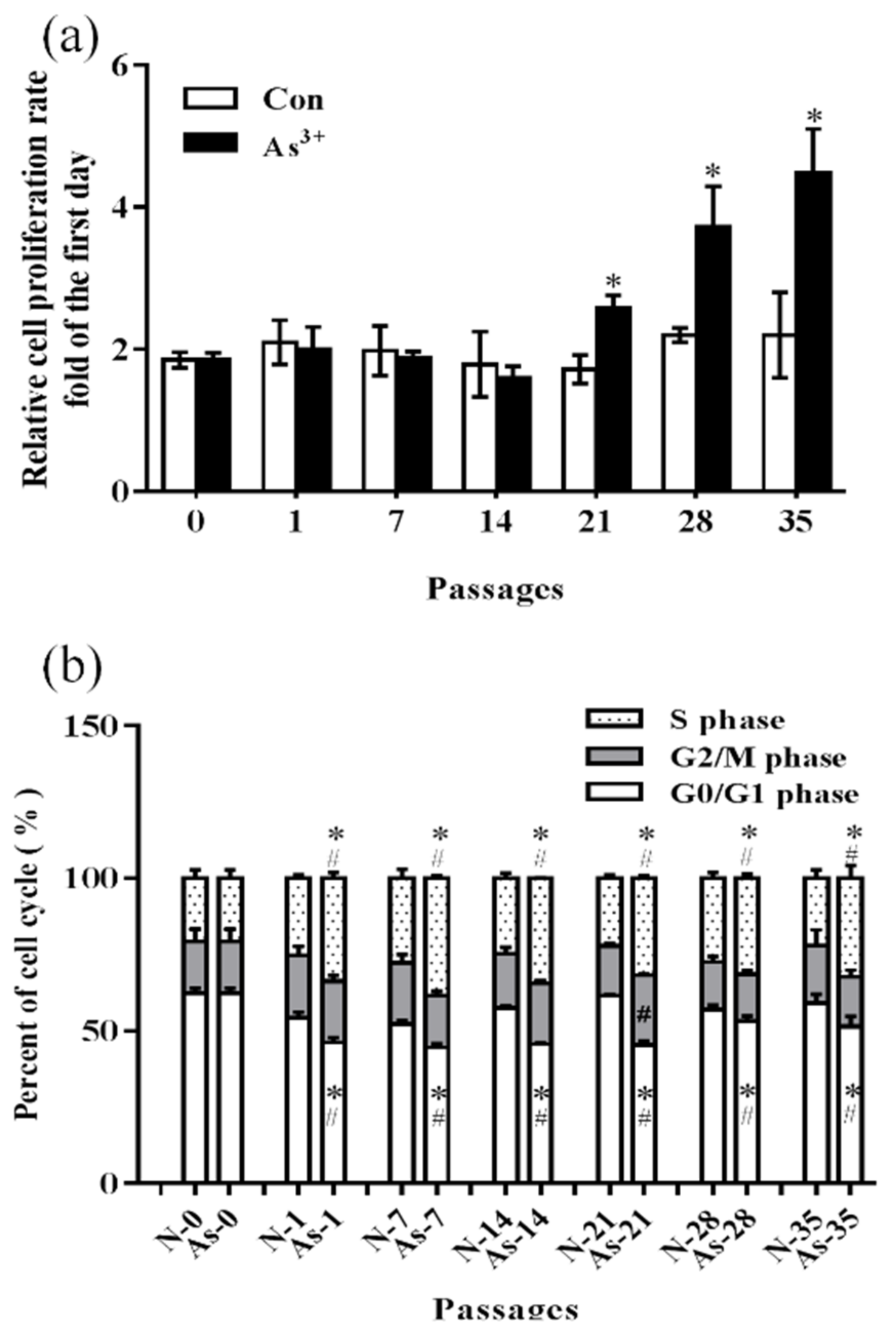
2.2. Proliferation of Cells Assay
2.3. Flow Cytometry Analysis of the Cell Cycle
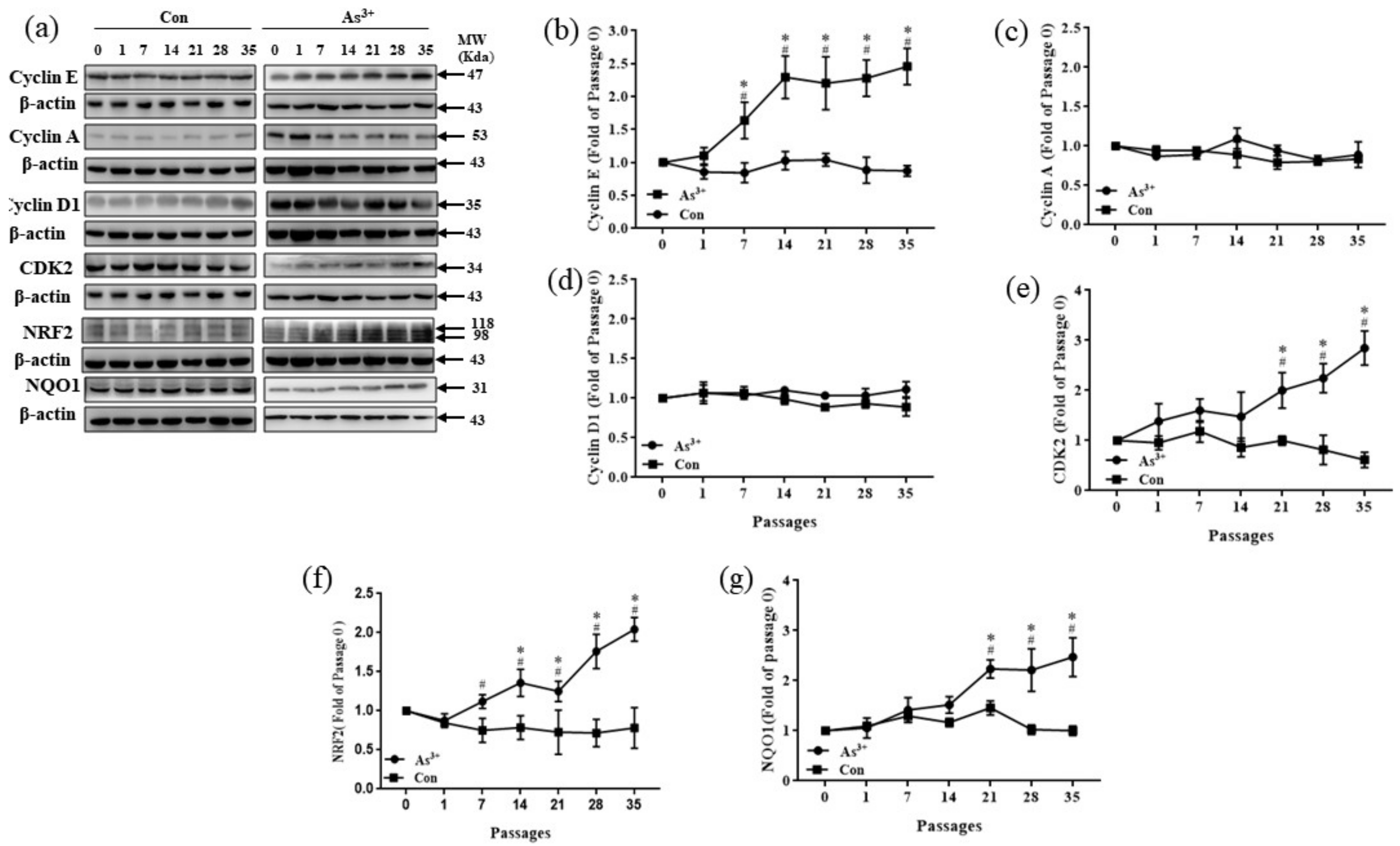
2.4. Western Blots
2.5. SiRNA Interference Assays
2.6. Wound-Healing (Cell Migration) Assay
2.7. Anchor-Independent Growth Experiment
2.8. Statistical Analysis
3. Results
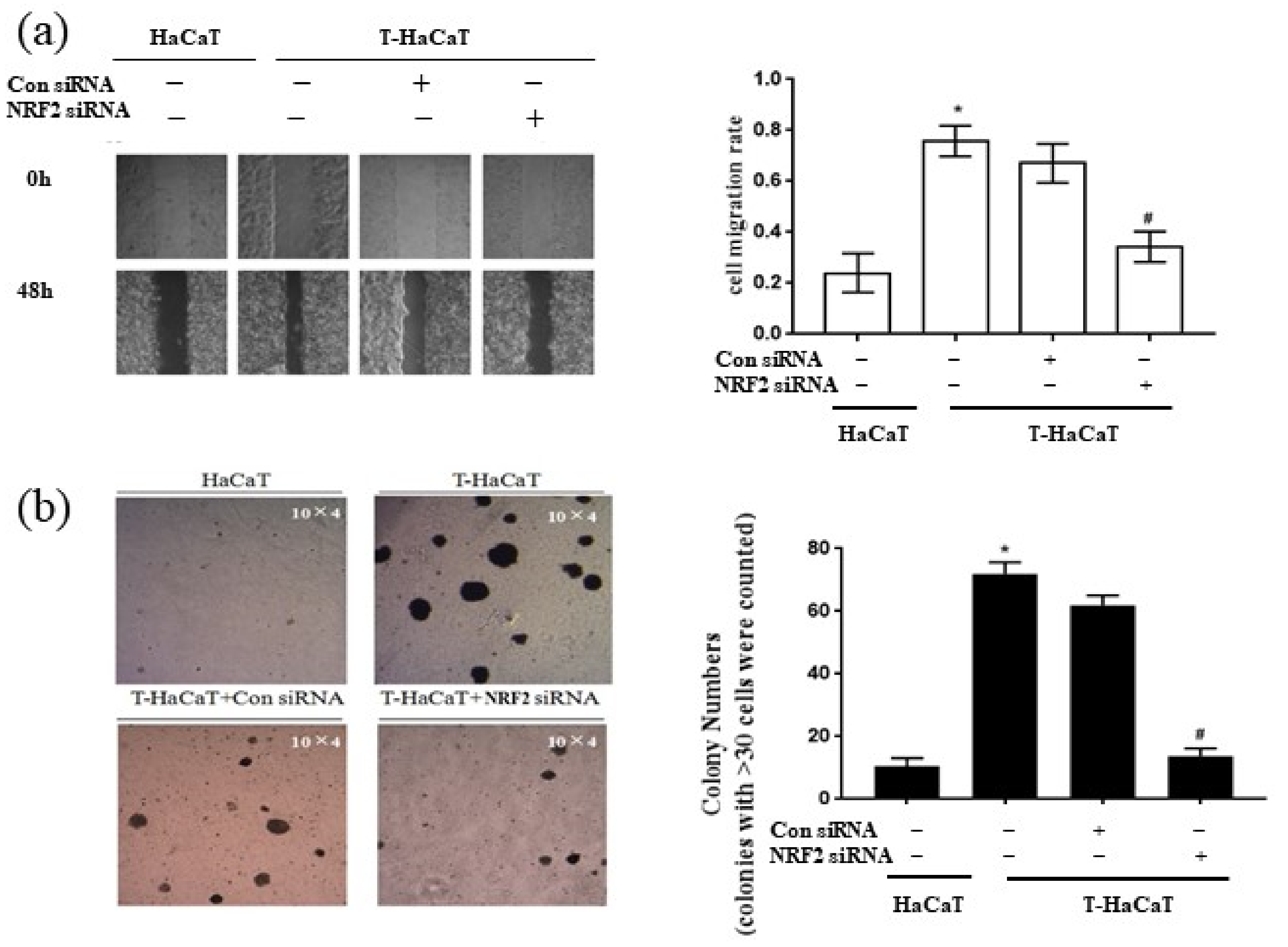
3.1. Malignant Phenotypes Were Induced in HaCaT Cells with Continuous Arsenic Exposure
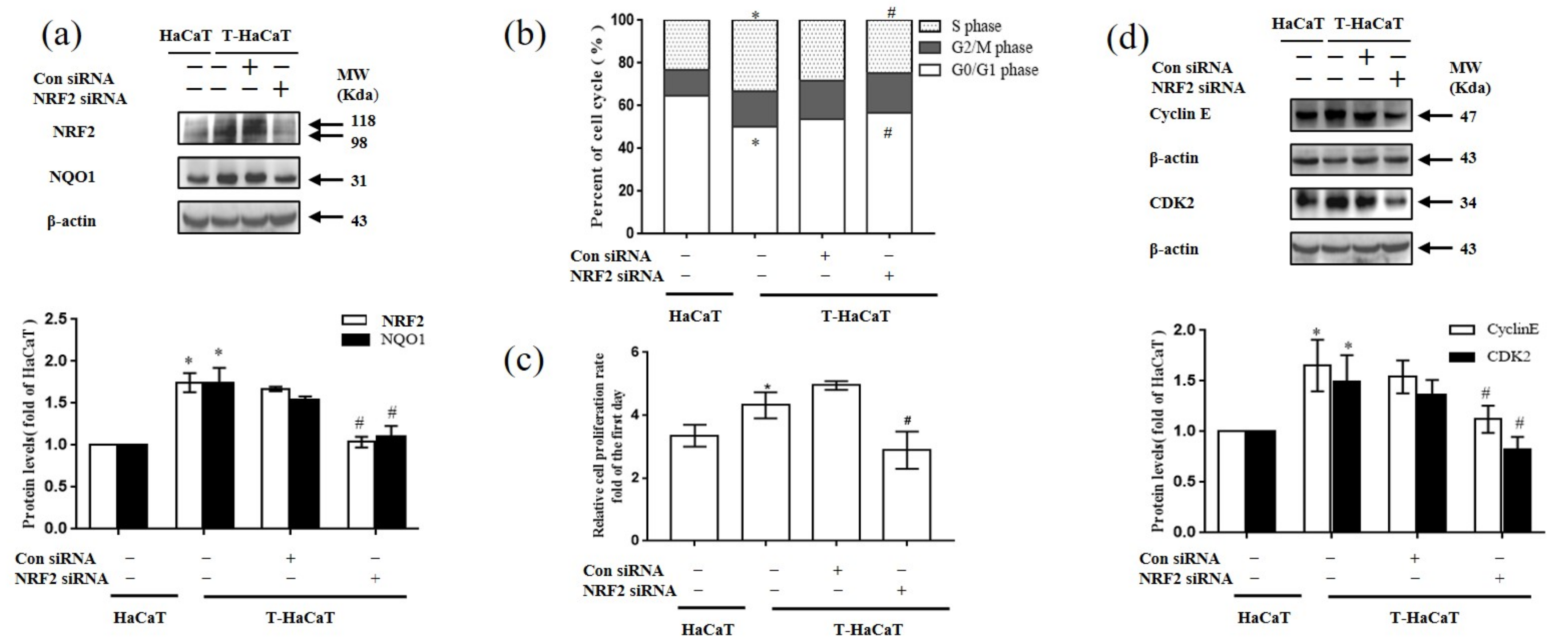
3.2. Decreased Cell Cycle-Related Proteins and NRF2 and NQO1 in HaCaT Cells Exposed to Arsenite
3.3. NRF2-Mediated Cellular Proliferation in Arsenite-Exposed HaCaT Cells
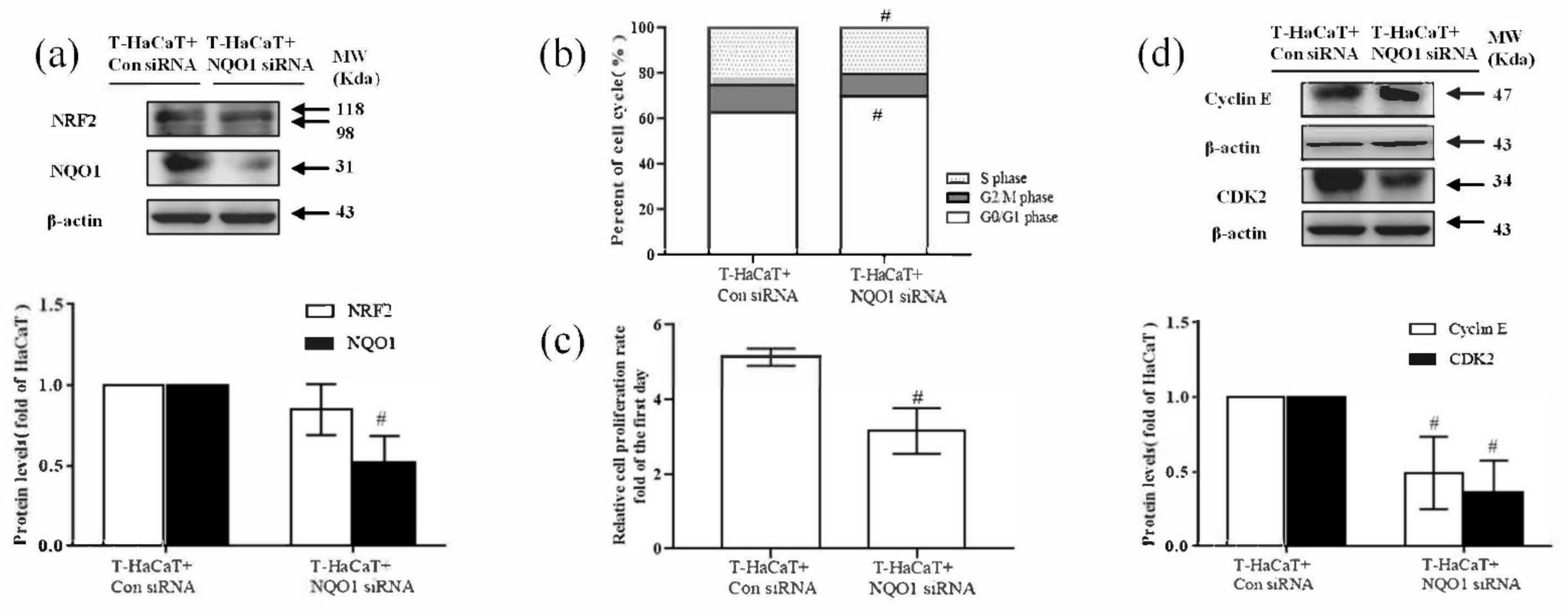
3.4. NQO1-Mediated Cellular Proliferation in Arsenite-Exposed HaCaT Cells
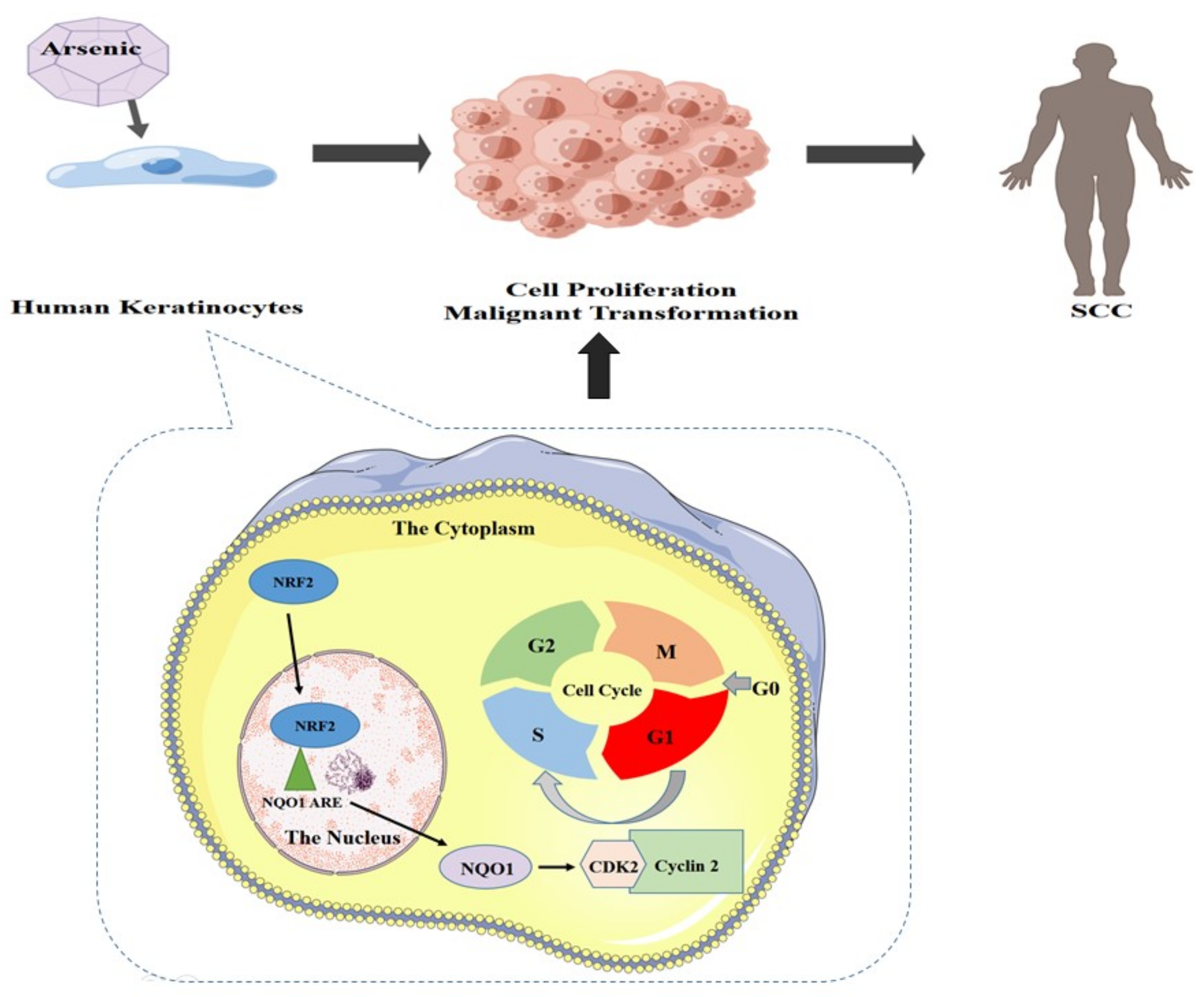
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| ARE | antioxidant response element |
| CDK2 | Cyclin-dependent kinase 2 |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | Fetal bovine serum |
| HaCaT | immortalized human keratinocyte cells |
| Keap1 | Kelch-like ECH-associated protein 1 |
| NaAsO2 | sodium arsenite |
| NRF2 | nuclear factor-erythroid-2 p45-related factor 2 |
| NQO-1 | NAD(P) H: Quinine oxidoreductase 1 |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| PBS | phosphate-buffered saline |
| PI | Propidium iodide |
| ROS | reactive oxygen species |
| T-HaCaT | arsenite-transformed HaCaT cells |
| HBE | human bronchial epithelial |
| MC | skin melanoma |
| NMSC | non-melanoma skin cancer |
| BCC | basal cell carcinoma |
| SCC | squamous cell carcinoma |
| UV | ultraviolet radiation |
| siRNA | small interfering RNA |
References
- Garelick, H.; Jones, H.; Dybowska, A.; Valsami-Jones, E. Arsenic pollution sources. Rev. Environ. Contam. Toxicol. 2008, 197, 17–60. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Song, K.H.; Chung, J.Y. Health effects of chronic arsenic exposure. J. Prev. Med. Public Health 2014, 47, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchounwou, P.B.; Wilson, B.; Ishaque, A. Important considerations in the development of public health advisories for arsenic and arsenic-containing compounds in drinking water. Rev. Environ. Health 1999, 14, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Rousselot, P.; Labaume, S.; Marolleau, J.P.; Larghero, J.; Noguera, M.H.; Brouet, J.C.; Fermand, J.P. Arsenic trioxide and melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer Res. 1999, 59, 1041–1048. [Google Scholar]
- Missimer, T.M.; Teaf, C.M.; Beeson, W.T.; Maliva, R.G.; Woolschlager, J.; Covert, D.J. Natural Background and Anthropogenic Arsenic Enrichment in Florida Soils, Surface Water, and Groundwater: A Review with a Discussion on Public Health Risk. Int. J. Environ. Res. Public Health 2018, 15, 2278. [Google Scholar] [CrossRef] [Green Version]
- Oremland, R.S.; Stolz, J.F. The ecology of arsenic. Science 2003, 300, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Hull, E.A.; Barajas, M.; Burkart, K.A.; Fung, S.R.; Jackson, B.P.; Barrett, P.M.; Neumann, R.B.; Olden, J.D.; Gawel, J.E. Human health risk from consumption of aquatic species in arsenic-contaminated shallow urban lakes. Sci. Total Environ. 2021, 770, 145318. [Google Scholar] [CrossRef]
- Xue, L.; Zhao, Z.; Zhang, Y.; Liao, J.; Wu, M.; Wang, M.; Sun, J.; Gong, H.; Guo, M.; Li, S.; et al. Dietary exposure to arsenic and human health risks in western Tibet. Sci. Total Environ. 2020, 731, 138840. [Google Scholar] [CrossRef]
- Monteiro, D.O.E.; Caixeta, E.S.; Santos, V.; Pereira, B.B. Arsenic exposure from groundwater: Environmental contamination, human health effects, and sustainable solutions. J. Toxicol. Environ. Health B Crit. Rev. 2021, 24, 119–135. [Google Scholar] [CrossRef]
- Byeon, E.; Kang, H.M.; Yoon, C.; Lee, J.S. Toxicity mechanisms of arsenic compounds in aquatic organisms. Aquat Toxicol. 2021, 237, 105901. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hasegawa, H.; Lim, R.P. Bioaccumulation, biotransformation and trophic transfer of arsenic in the aquatic food chain. Environ. Res. 2012, 116, 118–135. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Zhiqiang, G.; Dongdong, S.; Sen, D.; Li, Z. Arsenic speciation in wild marine organisms and a health risk assessment in a subtropical bay of China. Sci. Total Environ. 2018, 626, 621–629. [Google Scholar]
- Vahter, M.; Skroder, H.; Rahman, S.M.; Levi, M.; Derakhshani, H.J.; Kippler, M. Prenatal and childhood arsenic exposure through drinking water and food and cognitive abilities at 10 years of age: A prospective cohort study. Environ. Int. 2020, 139, 105723. [Google Scholar] [CrossRef]
- Mondal, D.; Rahman, M.M.; Suman, S.; Sharma, P.; Siddique, A.B.; Rahman, M.A.; Bari, A.; Kumar, R.; Bose, N.; Singh, S.K.; et al. Arsenic exposure from food exceeds that from drinking water in endemic area of Bihar, India. Sci. Total Environ. 2021, 754, 142082. [Google Scholar] [CrossRef]
- Joardar, M.; Das, A.; Chowdhury, N.R.; Mridha, D.; De, A.; Majumdar, K.K.; Roychowdhury, T. Health effect and risk assessment of the populations exposed to different arsenic levels in drinking water and foodstuffs from four villages in arsenic endemic Gaighata block, West Bengal, India. Environ. Geochem. Health 2021, 43, 3027–3053. [Google Scholar] [CrossRef]
- Baker, B.A.; Cassano, V.A.; Murray, C. Arsenic Exposure, Assessment, Toxicity, Diagnosis, and Management: Guidance for Occupational and Environmental Physicians. J. Occup. Environ. Med. 2018, 60, e634–e639. [Google Scholar] [CrossRef]
- Chou, C.H.; De Rosa, C.T. Case studies—Arsenic. Int. J. Hyg. Environ. Health 2003, 206, 381–386. [Google Scholar] [CrossRef]
- Jarup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Chakraborti, D.; Rahman, M.M.; Chatterjee, A.; Das, D.; Das, B.; Nayak, B.; Pal, A.; Chowdhury, U.K.; Ahmed, S.; Biswas, B.K.; et al. Fate of over 480 million inhabitants living in arsenic and fluoride endemic Indian districts: Magnitude, health, socio-economic effects and mitigation approaches. J Trace Elem Med Biol 2016, 38, 33–45. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Yedjou, C.G.; Udensi, U.K.; Pacurari, M.; Stevens, J.J.; Patlolla, A.K.; Noubissi, F.; Kumar, S. State of the science review of the health effects of inorganic arsenic: Perspectives for future research. Environ. Toxicol. 2019, 34, 188–202. [Google Scholar] [CrossRef]
- Pye-Smith, R.J. Arsenic Cancer, with Description of a Case. Proc. R. Soc. Med. 1913, 6, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Bungeler, W. Arsenic cancer. Munch. Med. Wochenschr. 1958, 100, 1117–1118. [Google Scholar]
- Wei, S.; Zhang, H.; Tao, S. A review of arsenic exposure and lung cancer. Toxicol. Res. 2019, 8, 319–327. [Google Scholar] [CrossRef]
- Mayer, J.E.; Goldman, R.H. Arsenic and skin cancer in the USA: The current evidence regarding arsenic-contaminated drinking water. Int. J. Dermatol. 2016, 55, e585–e591. [Google Scholar] [CrossRef]
- Jackson, R.; Grainge, J.W. Arsenic and cancer. Can. Med. Assoc. J. 1975, 113, 396–401. [Google Scholar]
- Hamilton, A. Industrial Toxicology, 2nd ed.; P. B. Hoeber: New York, NY, USA, 1949. [Google Scholar]
- Gordon, R. Skin cancer: An overview of epidemiology and risk factors. Semin. Oncol. Nurs. 2013, 29, 160–169. [Google Scholar] [CrossRef]
- Rosales-Castillo, J.A.; Acosta-Saavedra, L.C.; Torres, R.; Ochoa-Fierro, J.; Borja-Aburto, V.H.; Lopez-Carrillo, L.; Garcia-Vargas, G.G.; Gurrola, G.B.; Cebrian, M.E.; Calderon-Aranda, E.S. Arsenic exposure and human papillomavirus response in non-melanoma skin cancer Mexican patients: A pilot study. Int. Arch. Occup. Environ. Health 2004, 77, 418–423. [Google Scholar] [CrossRef]
- Surdu, S. Non-melanoma skin cancer: Occupational risk from UV light and arsenic exposure. Rev. Environ. Health 2014, 29, 255–264. [Google Scholar] [CrossRef]
- Surdu, S.; Fitzgerald, E.F.; Bloom, M.S.; Boscoe, F.P.; Carpenter, D.O.; Haase, R.F.; Gurzau, E.; Rudnai, P.; Koppova, K.; Vahter, M.; et al. Polymorphisms in DNA repair genes XRCC1 and XRCC3, occupational exposure to arsenic and sunlight, and the risk of non-melanoma skin cancer in a European case-control study. Environ. Res. 2014, 134, 382–389. [Google Scholar] [CrossRef]
- Kim, T.H.; Seo, J.W.; Hong, Y.S.; Song, K.H. Case-control study of chronic low-level exposure of inorganic arsenic species and non-melanoma skin cancer. J. Dermatol. 2017, 44, 1374–1379. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic. Biol. Med. 2015, 88, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geismann, C.; Arlt, A.; Sebens, S.; Schafer, H. Cytoprotection “gone astray”: Nrf2 and its role in cancer. OncoTargets Ther. 2014, 7, 1497–1518. [Google Scholar] [CrossRef] [Green Version]
- Montserrat, R.D.L.V.; Eli, C.; Donna, D.Z. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Kepa, J.K.; Winski, S.L.; Beall, H.D.; Anwar, A.; Siegel, D. NAD(P)H:quinone oxidoreductase 1 (NQO1): Chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. 2000, 129, 77–97. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, G.; Dong, B.; Piao, J.; Chen, L.; Lin, Z. The NQO1/PKLR axis promotes lymph node metastasis and breast cancer progression by modulating glycolytic reprogramming. Cancer Lett. 2019, 453, 170–183. [Google Scholar] [CrossRef]
- Butsri, S.; Kukongviriyapan, V.; Senggunprai, L.; Kongpetch, S.; Zeekpudsa, P.; Prawan, A. Downregulation of NAD(P)H:quinone oxidoreductase 1 inhibits proliferation, cell cycle and migration of cholangiocarcinoma cells. Oncol. Lett. 2017, 13, 4540–4548. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Li, J.; Martinka, M.; Li, G. The expression of NAD(P)H:quinone oxidoreductase 1 is increased along with NF-kappaB p105/p50 in human cutaneous melanomas. Oncol. Rep. 2010, 23, 973–979. [Google Scholar] [CrossRef]
- Wang, D.; Ma, Y.; Yang, X.; Xu, X.; Zhao, Y.; Zhu, Z.; Wang, X.; Deng, H.; Li, C.; Gao, F.; et al. Hypermethylation of the Keap1 gene inactivates its function, promotes Nrf2 nuclear accumulation, and is involved in arsenite-induced human keratinocyte transformation. Free Radic. Biol. Med. 2015, 89, 209–219. [Google Scholar] [CrossRef]
- Luo, S.; Lei, K.; Xiang, D.; Ye, K. NQO1 Is Regulated by PTEN in Glioblastoma, Mediating Cell Proliferation and Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 9146528. [Google Scholar] [CrossRef]
- Yang, X.; Wang, D.; Ma, Y.; Xu, X.; Zhu, Z.; Wang, X.; Deng, H.; Li, C.; Chen, M.; Tong, J.; et al. Continuous activation of Nrf2 and its target antioxidant enzymes leads to arsenite-induced malignant transformation of human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2015, 289, 231–239. [Google Scholar] [CrossRef]
- Tong, Y.H.; Zhang, B.; Yan, Y.Y.; Fan, Y.; Yu, J.W.; Kong, S.S.; Zhang, D.; Fang, L.; Su, D.; Lin, N.M. Dual-negative expression of Nrf2 and NQO1 predicts superior outcomes in patients with non-small cell lung cancer. Oncotarget 2017, 8, 45750–45758. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Deng, H.; Li, C.; Wang, X.; Shimoda, Y.; Tao, S.; Kato, K.; Zhang, J.; Yamanaka, K.; An, Y. Sustained high expression of NRF2 and its target genes induces dysregulation of cellular proliferation and apoptosis is associated with arsenite-induced malignant transformation of human bronchial epithelial cells. Sci. Total Environ. 2021, 756, 143840. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Schafer, K.A. The Cell Cycle: A Review. Vet. Pathol. 1998, 35, 461–478. [Google Scholar] [CrossRef]
- Yu, S.; Liao, W.T.; Lee, C.H.; Chai, C.Y.; Yu, C.L.; Yu, H.S. Immunological dysfunction in chronic arsenic exposure: From subclinical condition to skin cancer. J. Dermatol. 2018, 45, 1271–1277. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Smith, E.; Goodall, G.J.; Drew, P.A.; Brabletz, T.; Yang, C. Reversal and prevention of arsenic-induced human bronchial epithelial cell malignant transformation by microRNA-200b. Toxicol. Sci. 2011, 121, 110–122. [Google Scholar] [CrossRef]
- Ge, Y.; Zhu, J.; Wang, X.; Zheng, N.; Tu, C.; Qu, J.; Ren, X. Mapping dynamic histone modification patterns during arsenic-induced malignant transformation of human bladder cells. Toxicol. Appl. Pharmacol. 2018, 355, 164–173. [Google Scholar] [CrossRef]
- Benbrahim-Tallaa, L.; Waterland, R.A.; Styblo, M.; Achanzar, W.E.; Webber, M.M.; Waalkes, M.P. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: Aberrant genomic DNA methylation and K-ras oncogene activation. Toxicol. Appl. Pharmacol. 2005, 206, 288–298. [Google Scholar] [CrossRef]
- Hunt, K.M.; Srivastava, R.K.; Elmets, C.A.; Athar, M. The mechanistic basis of arsenicosis: Pathogenesis of skin cancer. Cancer Lett. 2014, 354, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Rusanov, A.L.; Romashin, D.D.; Zgoda, V.G.; Butkova, T.V.; Luzgina, N.G. Protein dataset of immortalized keratinocyte HaCaT cells and normal human keratinocytes. Data Brief. 2021, 35, 106871. [Google Scholar] [CrossRef]
- Valerio, H.P.; Ravagnani, F.G.; Ronsein, G.E.; Di Mascio, P. A single dose of Ultraviolet-A induces proteome remodeling and senescence in primary human keratinocytes. Sci. Rep. 2021, 11, 23355. [Google Scholar] [CrossRef]
- Lee, R.H.; Oh, J.D.; Hwang, J.S.; Lee, H.K.; Shin, D. Antitumorigenic effect of insect-derived peptide poecilocorisin-1 in human skin cancer cells through regulation of Sp1 transcription factor. Sci. Rep. 2021, 11, 18445. [Google Scholar] [CrossRef]
- Bicheng, F.; Pradeep, D.; Fengjie, L.; Laura, G.; Martina, B.; Alfonso, M.; Deniz, T. Pyrenosetin D, a New Pentacyclic Decalinoyltetramic Acid Derivative from the Algicolous Fungus Pyrenochaetopsis sp. FVE-087. Mar. Drugs 2020, 18, 281. [Google Scholar]
- Makoto, S.; Kazunori, H.; Masafumi, Y.; Mohammad, D.A.; Said, H.F.; Nobuyuki, H.; Lisa, K.; Kiyoshi, Y.; Masashi, K. Lithium promotes malignant transformation of nontumorigenic cells in vitro. Sci. Total Environ. 2020, 744, 140830. [Google Scholar]
- Wright, C.; Iyer, A.K.V.; Wang, L.; Wu, N.; Yakisich, J.S.; Rojanasakul, Y.; Azad, N. Effects of titanium dioxide nanoparticles on human keratinocytes. Drug Chem. Toxicol. 2017, 40, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.S.; Zhang, Z.G.; Du, G.Y.; Sun, H.L.; Liu, H.Y.; Zhou, Z.; Gou, X.M.; Wu, X.H.; Yu, X.Y.; Huang, Y.H. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1alpha/Notch1 axis. J. Cell Mol. Med. 2019, 23, 3451–3463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Jiang, L.; Yang, Y.; Wu, M.; Liu, B.; Shi, Y.; Shen, Q.; Jiang, X.; He, Y.; Cheng, D.; et al. PAQR4 promotes chemoresistance in non-small cell lung cancer through inhibiting Nrf2 protein degradation. Theranostics 2020, 10, 3767–3778. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Homma, S.; Ishii, Y.; Morishima, Y.; Yamadori, T.; Matsuno, Y.; Haraguchi, N.; Kikuchi, N.; Satoh, H.; Sakamoto, T.; Hizawa, N.; et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 2009, 15, 3423–3432. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Yamadori, T.; Ishii, Y.; Homma, S.; Morishima, Y.; Kurishima, K.; Itoh, K.; Yamamoto, M.; Minami, Y.; Noguchi, M.; Hizawa, N. Molecular mechanisms for the regulation of Nrf2-mediated cell proliferation in non-small-cell lung cancers. Oncogene 2012, 31, 4768–4777. [Google Scholar] [CrossRef] [Green Version]
- Hosein, A.N.; Beg, M.S. Pancreatic Cancer Metabolism: Molecular Mechanisms and Clinical Applications. Curr. Oncol. Rep. 2018, 20, 56. [Google Scholar] [CrossRef] [PubMed]
- Marco, G.; Aijaz, A.W.; Gang, L. The NAD(P)H:Quinone Oxidoreductase 1 induces cell cycle progression and proliferation of melanoma cells. Free Radical. Bio. Med. 2010, 48, 1601–1609. [Google Scholar]
- Marrot, L.; Jones, C.; Perez, P.; Meunier, J.R. The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment. Cell Melanoma Res. 2008, 21, 79–88. [Google Scholar] [CrossRef]
- Yang, Y.; Zheng, J.; Wang, M.; Zhang, J.; Tian, T.; Wang, Z.; Yuan, S.; Liu, L.; Zhu, P.; Gu, F.; et al. NQO1 promotes an aggressive phenotype in hepatocellular carcinoma via amplifying ERK-NRF2 signaling. Cancer Sci. 2021, 112, 641–654. [Google Scholar] [CrossRef]
- Cedric, O.R.; Panos, G.Z.; Dionysios, V.C.; Dionysios, V.C.; Massimo, B.; Gerasimos, P.S. Keap1/Nrf2 Signaling: A New Player in Thyroid Pathophysiology and Thyroid Cancer. Front. Endocrinol. 2019, 10, 510. [Google Scholar]
- Carpenter, E.L.; Becker, A.L.; Indra, A.K. NRF2 and Key Transcriptional Targets in Melanoma Redox Manipulation. Cancers 2022, 14, 1531. [Google Scholar] [CrossRef]
- Chu, C.; Geng, Y.; Zhou, Y.; Sicinski, P. Cyclin E in normal physiology and disease states. Trends Cell Biol. 2021, 31, 732–746. [Google Scholar] [CrossRef]
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An integrated view of cyclin E function and regulation. Cell Cycle 2012, 11, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Lee, C.; An, H.; Kang, H.C.; Lee, H.S.; Lee, J.Y.; Oh, S.; Cho, S.; Kim, D.J.; Cho, Y. Fargesin Inhibits EGF-Induced Cell Transformation and Colon Cancer Cell Growth by Suppression of CDK2/Cyclin E Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 2073. [Google Scholar] [CrossRef]
- Liang, Y.; Gao, H.; Lin, S.Y.; Goss, J.A.; Brunicardi, F.C.; Li, K. siRNA-based targeting of cyclin E overexpression inhibits breast cancer cell growth and suppresses tumor development in breast cancer mouse model. PLoS ONE 2010, 5, e12860. [Google Scholar] [CrossRef]
- Lastra, D.; Escoll, M.; Cuadrado, A. Transcription Factor NRF2 Participates in Cell Cycle Progression at the Level of G1/S and Mitotic Checkpoints. Antioxidants 2022, 11, 946. [Google Scholar] [CrossRef]
- Reddy, N.M.; Kleeberger, S.R.; Bream, J.H.; Fallon, P.G.; Kensler, T.W.; Yamamoto, M.; Reddy, S.P. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene 2008, 27, 5821–5832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a Therapeutic and Diagnostic Target in Cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef]
- Oh, E.T.; Park, H.J. Implications of NQO1 in cancer therapy. BMB Rep. 2015, 48, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Spagnuolo, C.; Russo, G.L.; Skalicka-Wozniak, K.; Daglia, M.; Sobarzo-Sanchez, E.; Nabavi, S.F.; Nabavi, S.M. Nrf2 targeting by sulforaphane: A potential therapy for cancer treatment. Crit. Rev. Food Sci. Nutr. 2018, 58, 1391–1405. [Google Scholar] [CrossRef]
- Hammad, A.; Namani, A.; Elshaer, M.; Wang, X.J.; Tang, X. “NRF2 addiction” in lung cancer cells and its impact on cancer therapy. Cancer Lett. 2019, 467, 40–49. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, I.; Cao, M.; Su, Z.Y.; Wu, R.; Guo, Y.; Fang, M.; Kong, A.N. Fucoxanthin Elicits Epigenetic Modifications, Nrf2 Activation and Blocking Transformation in Mouse Skin JB6 P+ Cells. AAPS J. 2018, 20, 32. [Google Scholar] [CrossRef]
- Chun, K.S.; Kundu, J.; Kundu, J.K.; Surh, Y.J. Targeting Nrf2-Keap1 signaling for chemoprevention of skin carcinogenesis with bioactive phytochemicals. Toxicol. Lett. 2014, 229, 73–84. [Google Scholar] [CrossRef]
- Yang, H.L.; Lee, C.L.; Korivi, M.; Liao, J.W.; Rajendran, P.; Wu, J.J.; Hseu, Y.C. Zerumbone protects human skin keratinocytes against UVA-irradiated damages through Nrf2 induction. Biochem. Pharmacol. 2018, 148, 130–146. [Google Scholar] [CrossRef]
- Sivinski, J.; Zhang, D.D.; Chapman, E. Targeting NRF2 to treat cancer. Semin. Cancer Biol. 2021, 76, 61–73. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, R.; Wu, R.; Ramirez, C.N.; Sargsyan, D.; Li, S.; Wang, L.; Cheng, D.; Wang, C.; Hudlikar, R.; et al. DNA methylome and transcriptome alterations and cancer prevention by triterpenoid ursolic acid in UVB-induced skin tumor in mice. Mol. Carcinog. 2019, 58, 1738–1753. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Yan, R.; Mo, Y.; Xia, H.; Deng, H.; Wang, X.; Li, C.; Kato, K.; Zhang, H.; Jin, T.; et al. The Potential Key Role of the NRF2/NQO1 Pathway in the Health Effects of Arsenic Pollution on SCC. Int. J. Environ. Res. Public Health 2022, 19, 8118. https://doi.org/10.3390/ijerph19138118
Yang Q, Yan R, Mo Y, Xia H, Deng H, Wang X, Li C, Kato K, Zhang H, Jin T, et al. The Potential Key Role of the NRF2/NQO1 Pathway in the Health Effects of Arsenic Pollution on SCC. International Journal of Environmental Research and Public Health. 2022; 19(13):8118. https://doi.org/10.3390/ijerph19138118
Chicago/Turabian StyleYang, Qianlei, Rui Yan, Yuemei Mo, Haixuan Xia, Hanyi Deng, Xiaojuan Wang, Chunchun Li, Koichi Kato, Hengdong Zhang, Tingxu Jin, and et al. 2022. "The Potential Key Role of the NRF2/NQO1 Pathway in the Health Effects of Arsenic Pollution on SCC" International Journal of Environmental Research and Public Health 19, no. 13: 8118. https://doi.org/10.3390/ijerph19138118
APA StyleYang, Q., Yan, R., Mo, Y., Xia, H., Deng, H., Wang, X., Li, C., Kato, K., Zhang, H., Jin, T., Zhang, J., & An, Y. (2022). The Potential Key Role of the NRF2/NQO1 Pathway in the Health Effects of Arsenic Pollution on SCC. International Journal of Environmental Research and Public Health, 19(13), 8118. https://doi.org/10.3390/ijerph19138118