The Effects of Arsenic Trioxide on DNA Synthesis and Genotoxicity in Human Colon Cancer Cells


 and
and 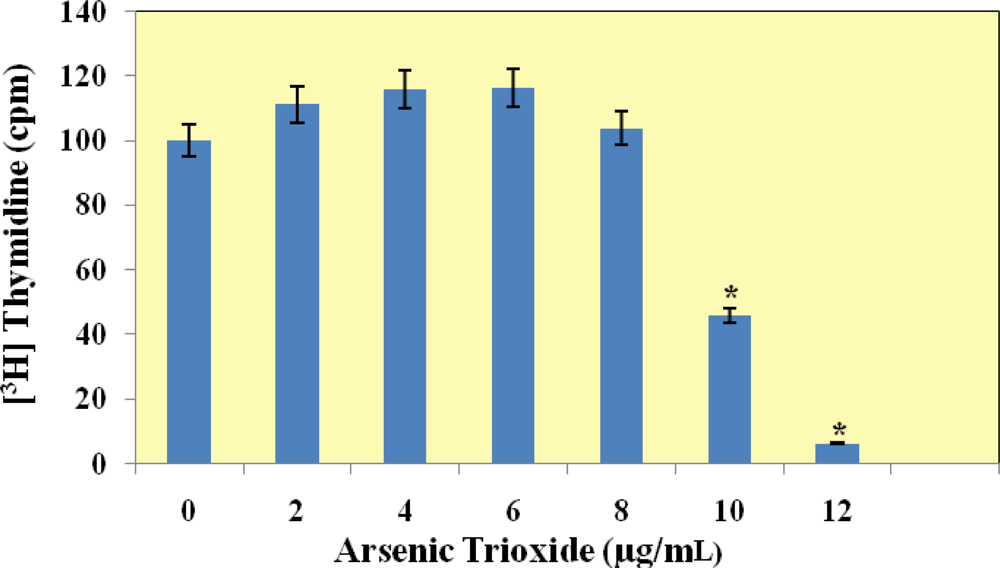{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
Reagents and Cell Line
Cell Culture
[3H]Thymidine Incorporation Assay
Genotoxicity Study
Statistical Analysis
[3H]Thymidine Incorporation Assay
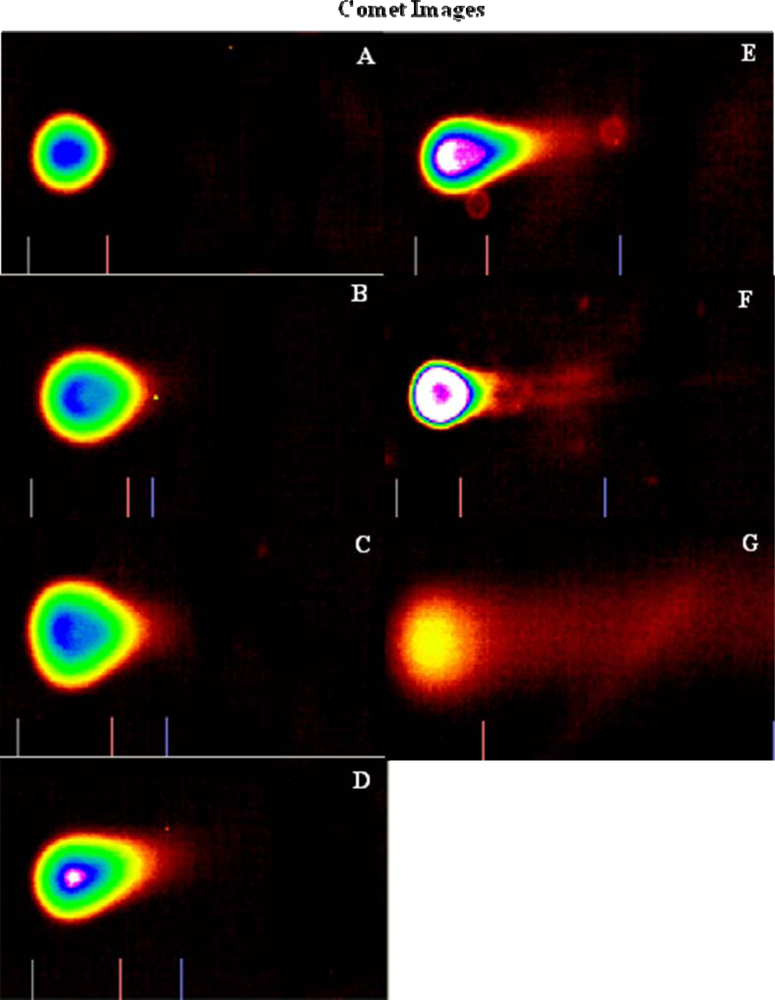
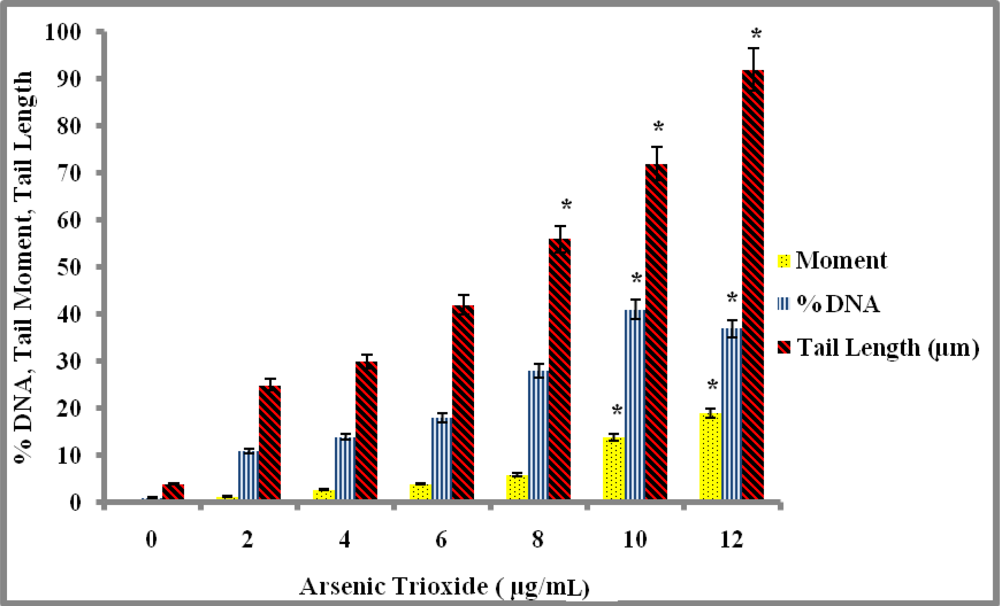
Genotoxicity Study
3. Results
[3H]Thymidine Incorporation Assay
Genotoxicity Study
4. Discussion
5. Conclusions
Acknowledgments
References
- Jermal, A; Siegel, R; Ward, E; Hao, Y; Xu, J; Thun, MJ. Cancer Statistics, 2009. CA Cancer J. Clin 2009, 59, 225–249. [Google Scholar]
- Tapio, S; Grosche, B. Arsenic in the aetiology of cancer. Mutat. Res 2006, 612, 215–246. [Google Scholar]
- Ghosh, P; Banerjee, M; Giri, AK; Ray, K. Toxicogenomics of arsenic: classical ideas and recent advances. Mutat. Res 2008, 659, 293–301. [Google Scholar]
- Tchounwou, PB; Patlolla, AK; Centeno, JA. Carcinogenic and systemic health effects associated with arsenic exposure—a critical review. Toxicol Pathol 2003, 31, 575–588. [Google Scholar]
- IARC. Monographs on the Evaluation of Carcinogenic Risks of Chemcials to Humans Supplement F Overall Evaluation of Carcinogenicity; International Agency for Research on Cancer, World Health Organization: Lyon, France, 1987; pp. 29–55. [Google Scholar]
- Tchounwou, PB; Centeno, JA; Patlolla, AK. Arsenic toxicity, mutagenesis and carcinogenesis: a health risk assessment and management approach. Mol. Cell Biochem 2004, 255, 47–55. [Google Scholar]
- Tchounwou, PB; Wilson, B; Ishaque, A. Important considerations in the development of public health advisories for arsenic and arsenic-containing compounds in drinking water. Rev. Environ. Health 1999, 19, 133–143. [Google Scholar]
- Roy, P; Saha, A. Metabolism and toxicity of arsenic: A human carcinogen. Curr. Sci 2002, 82, 38–45. [Google Scholar]
- Abernathy, C; Liu, Y; Longfellow, D; Aposhian, V; Beck, B; Fowler, B; Goyer, R; Menzer, R; Rossman, T; Thompson, C; Waalkes, M. Arsenic: health effects, mechanism of actions, and research issues. Environ. Health Perspect 1999, 107, 1–8. [Google Scholar]
- Kitchin, KT. Recent advances in arsenic carcinogenesis: Modes of action, Animal Model Systems, and Methylated arsenic metabolites. Toxicol. Appl. Pharmacol 2001, 172, 249–261. [Google Scholar]
- Kligerman, AD; Doerr, CL; Tennant, AH; Barrington-Brock, K; Allen, JW; Winkfield, E; Poorman-Allen, P; Kundu, B; Funasaka, K; Roop, BC; Mass, MJ; DeMarini, DM. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: induction of chromosomal mutations but not gene mutations. Environ. Mol. Mutagen 2003, 42, 192–205. [Google Scholar]
- Galanis, A; Karapetsas, A; Sandaltzopoulos, R. Metal-induced carcinogenesis, oxidative stress and hypoxia signaling. Mutat. Res 2009, 674, 31–35. [Google Scholar]
- Hughes, MF. Arsenic toxicity and potential mechanisms of action. Toxicol. Lett 2002, 122, 1–16. [Google Scholar]
- Miller, WH; Schipper, HM; Lee, JS; Waxman, S. Mechanisms of action of arsenic trioxide. Cancer Res 2002, 62, 3893–3903. [Google Scholar]
- Frumkin, H; Thun, MJ. Arsenic. CA J. Clin 2001, 51, 254–262. [Google Scholar]
- Guo, H. Cancer risks associated with arsenic in drinking water. Environ. Health Perspect 2007, 115, A339–A340. [Google Scholar]
- Yoshida, T; Yamauchi, H; Fan, Sun, G. Chronic health effects in people exposed to arsenic via the drinking water: dose-response relationships in review. Toxicol Appl Pharmacol 2004, 198, 243–252. [Google Scholar]
- NRC (National Research Council). Arsenic in Drinking Water: Update; National Academy Press: Washington, DC, USA, 1999. [Google Scholar]
- NRC (National Research Council). Arsenic in Drinking Water: Update; National Academy Press: Washington, DC, USA, 2001. [Google Scholar]
- Klein, CB; Leszczynska, J; Hickey, C; Rossman, TG. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol. Appl. Pharmacol 2007, 222, 289–297. [Google Scholar]
- Patlolla, A; Tchounwou, PB. Cytogenetic evaluation of arsenic trioxide toxicity in Sprague-Dawley rats. Mutat. Res 2005, 587, 126–133. [Google Scholar]
- Hartman, A; Speit, G. Comparative investigations of the Genotoxic effects of metals in the single gelassay and the sister-chromatid exchange tests. Environ. Mol. Mutagen 1994, 23, 299–305. [Google Scholar]
- Jha, AN; Noditi, M; Nilsson, R; Natarajan, AT. Genotoxic effects of sodium arsenite on human cell. Mutat. Res 1992, 284, 215–221. [Google Scholar]
- Hei, T; Filipic, M. Role of Oxidative damage in the genotoxicity of arsenic. Free Radic Bio Med 2004, 37, 574–581. [Google Scholar]
- Graham-Evans, B; Tchounwou, PB; Cohly, HHP. Cytotoxicity and proliferation studies with arsenic in established human cell lines: Keratinocytes, melanocytes, dendritic cells, dermal fibroblasts, microvascular endothethial cells, monocytes and T-cells. Int. J. Mol. Sci 2003, 4, 13–21. [Google Scholar]
- Gradecla, D; Paris, J; Waspwocz, W. Selected mechanisms of genotoxic effects of inorganic arsenic compounds. Int. J. Occup. Med. Environ. Health 2001, 14, 317–328. [Google Scholar]
- Hartwig, A; Asmuss, M; Ehleben, I; Herzer, H; Kostelac, D; Pelzer, A; Schwerdtle, T; Burkle, A. Interference by Toxic Metal Ions with DNA repair processes and cell cycle control: molecular mechanisms. Environ. Health Perspect 2002, 110, 797–799. [Google Scholar]
- Huang, C; Ke, Q; Costa, M; Shi, X. Molecular mechanisms of arsenic carcinogenesis. Mol. Cell Biochem 2004, 255, 57–66. [Google Scholar]
- Dong, Z. The molecular mechanisms of arsenic-induced cell transformation and apoptosis. Environ. Health Perspect 2002, 110, 757–759. [Google Scholar]
- Lynn, S; Gurr, JR; Lai, HT; Jan, KY. NADH oxidase activation is involved in arsenite induced oxidative DNA damage in human vascular smooth muscle cells. Circ. Res 2000, 86, 514–519. [Google Scholar]
- Nakamuro, K; Sayato, Y. Comparative studies of chromosomal aberration induced by trivalent and pentavalent arsenic. Mutat. Res 1981, 88, 73–80. [Google Scholar]
- Li, JH; Rossman, TG. Inhibition of DNA ligase activity by arsenite: a possible mechanism of its comutagenesis. Mol. Toxicol 1989, 2, 1–9. [Google Scholar]
- Murgo, AJ. Clinical trials of arsenic trioxide in hematologic and solid tumors: overview of the National Cancer Institute Cooperative Research and Development Studies. Oncologist 2001, 6, 22–28. [Google Scholar]
- Chen, GQ; Zhu, J; Shi, XG; Ni, HU; Zhong, HJ; Si, GY; Jin, XL; Tang, W; Li, XS; Xong, SM; Shen, ZX; Sun, GL; Ma, J; Zhang, P; Zhang, TD; Gazin, C; Naoe, T; Chen, ST; Wang, ZY; Chen, Z. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: arsenic trioxide induces NB 4 cell apoptosis with down-regulation of bcl-2 expression and modulation of PML-RAR α/PML proteins. Blood 1996, 88, 1052–1061. [Google Scholar]
- Hu, J; Liu, YF; Wu, CF; Xu, F; Shen, ZX; Zhu, YM; Li, JM; Tang, W; Zhao, WL; Wu, W; Sun, HP; Chen, QS; Chen, B; Zhou, GB; Zelent, A; Waxman, S; Wang, ZY; Chen, SJ; Chen, Z. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3342–3347. [Google Scholar]
- Munshi, N. Arsenic trioxide: an emerging therapy for multiple myeloma. Oncologist 2001, 6, 17–21. [Google Scholar]
- Monzen, H; Griffin, RJ; Williams, BW; Amano, M; Ando, S; Hasegawa, T. Study of Arsenic Trioxide-induced Vascular Shutdown and Enhancement with Radiation in solid tumor. Radiation Medicine 2004, 22, 205–211. [Google Scholar]
- Maeda, H; Hori, S; Ohizumi, H; Segawa, T; Kakehi, Y; Ogawa, O; Kakizuka, A. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death Differentiation 2004, 11, 737–746. [Google Scholar]
- Tice, RR; Agurell, E; Anderson, D; Burlinson, B; Hartmann, A; Kobayashi, H; Miyamae, Y; Ryu, J; Sasaki, YF. Single Cell Gel/Comet Assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen 2000, 35, 206–221. [Google Scholar]
- Huang, Y; Zhang, J; McHenry, KT; Kim, MM; Zeng, W; Lopez-Pajares, V; Dibble, CC; Mizgerd, JP; Yuan, ZM. Induction of cytoplasmic accumulation of p53: a mechanism for low levels of arsenic exposure to predispose cells for malignant transformation. Cancer Res 2008, 68, 9131–9136. [Google Scholar]
- Stevens, JJ; Graham-Evans, B; Walker, AM; Tchounwou, PB. Cytotoxic effect of arsenic trioxide in adenocarcinoma colorectal cancer (HT-29) cells. Metal Ions in Biology and Medicine 2008, 10, 458–462. [Google Scholar]
- Tchounwou, PB; Wilson, BA; Abdelghani, AA; Ishaque, AB; Patlolla, AK. Differential cytotoxicity and gene expression in human liver carcinoma (HepG2) cells exposed to arsenic trioxide, and monosodium acid methanearsonate (MSMA). Int. J. Mol. Sci 2002, 3, 1117–1132. [Google Scholar]
- Yedjou, CG; Tchouwou, PB. In-vitro cytotoxic and genotoxic effects of arsenic trioxide on human leukemia (HL-60) cells using the MTT and alkaline single cell electrophoresis (Comet) assays. Mole. Cell Biochem 2007, 301, 123–130. [Google Scholar]
- Walker, AM; Stevens, JJ; Tchounwou, PB. Arsenic trioxide mediated cytotoxicity and cell proliferation in breast and lung carcinoma cell lines. Metal Ions in Biology and Medicine 2006, 9, 287–292. [Google Scholar]
- Ye, J; Li, A; Liu, Q; Wang, X; Zhou, J. Inhibition of mitogen-activated protein kinase, kinase enhances apoptosis induced by arsenic trioxide in human breast cancer MCF-7 cells. Clin. Exp. Pharmacol. Physiol 2005, 32, 1042–1048. [Google Scholar]
- Hanadeh, HK; Trouba, KL; Amin, RP; Afshari, CA; Germolec, D. Coordination of altered DNA repair and damage pathways in arsenite-exposed keratinocytes. Toxicol. Sci 2002, 69, 306–316. [Google Scholar]
- Germolec, DR; Yoshida, T; Gaido, K; Wilmer, JL; Simeonova, PP; Kayama, F; Burleson, F; Dong, W; Lange, RW; Luster, MI. Arsenic induces overexpression of growth factors in human keratinocytes. Toxicol. Appl. Pharmacol 1996, 141, 309–318. [Google Scholar]
- Trouba, KJ; Wauson, EM; Vorce, RL. Sodium arsenite-induced dysregulation of proteins involved in proliferative signaling. Toxicol. Appl. Pharmacol 2000, 164, 161–170. [Google Scholar]
- Lee, HR; Cheong, J; Kim, SJ; Lee, NS; Park, HS; Won, JH. Sulindac enhances arsenic trioxide-mediated apotosis by inhibition of NF-kappaB in HCT116 colon cancer cells. Oncol. Rep 2008, 20, 41–47. [Google Scholar]
- Kent, CR; Eady, JJ; Ross, GM; Steel, GG. The comet moment as a measure of DNA damage in the comet assay. Int. J. Radiat. Biol 1995, 67, 655–660. [Google Scholar]
- Dopp, E; von Recklinghausen, U; Hartmann, LM; Stueckradt, I; Polllok, I; Rabieh, S; Hao, L; Nussler, A; Katier, C; Hirner, AV; Rettenmeier, AW. Subcellular distribution of inorganic and methylated arsenic compounds in human urothelial cells and human hepatocytes. Drug Metab. Disposition 2008, 5, 971–979. [Google Scholar]
- Guillamet, E; Creus, A; Ponti, J; Sabbioni, E; Fortaner, S; Marcos, R. In vitro DNA damage by arsenic compounds in a human lymphoblastoid cell line (TK6) assessed by the alkaline Comet assay. Mutagenesis 2004, 19, 129–135. [Google Scholar]



© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stevens, J.J.; Graham, B.; Walker, A.M.; Tchounwou, P.B.; Rogers, C. The Effects of Arsenic Trioxide on DNA Synthesis and Genotoxicity in Human Colon Cancer Cells. Int. J. Environ. Res. Public Health 2010, 7, 2018-2032. https://doi.org/10.3390/ijerph7052018
Stevens JJ, Graham B, Walker AM, Tchounwou PB, Rogers C. The Effects of Arsenic Trioxide on DNA Synthesis and Genotoxicity in Human Colon Cancer Cells. International Journal of Environmental Research and Public Health. 2010; 7(5):2018-2032. https://doi.org/10.3390/ijerph7052018
Chicago/Turabian StyleStevens, Jacqueline J., Barbara Graham, Alice M. Walker, Paul B. Tchounwou, and Christian Rogers. 2010. "The Effects of Arsenic Trioxide on DNA Synthesis and Genotoxicity in Human Colon Cancer Cells" International Journal of Environmental Research and Public Health 7, no. 5: 2018-2032. https://doi.org/10.3390/ijerph7052018
APA StyleStevens, J. J., Graham, B., Walker, A. M., Tchounwou, P. B., & Rogers, C. (2010). The Effects of Arsenic Trioxide on DNA Synthesis and Genotoxicity in Human Colon Cancer Cells. International Journal of Environmental Research and Public Health, 7(5), 2018-2032. https://doi.org/10.3390/ijerph7052018