Different Length (DL) qPCR for Quantification of Cell Killing by UV-induced DNA Damage

Abstract
:1. Introduction
2. Experimental Section
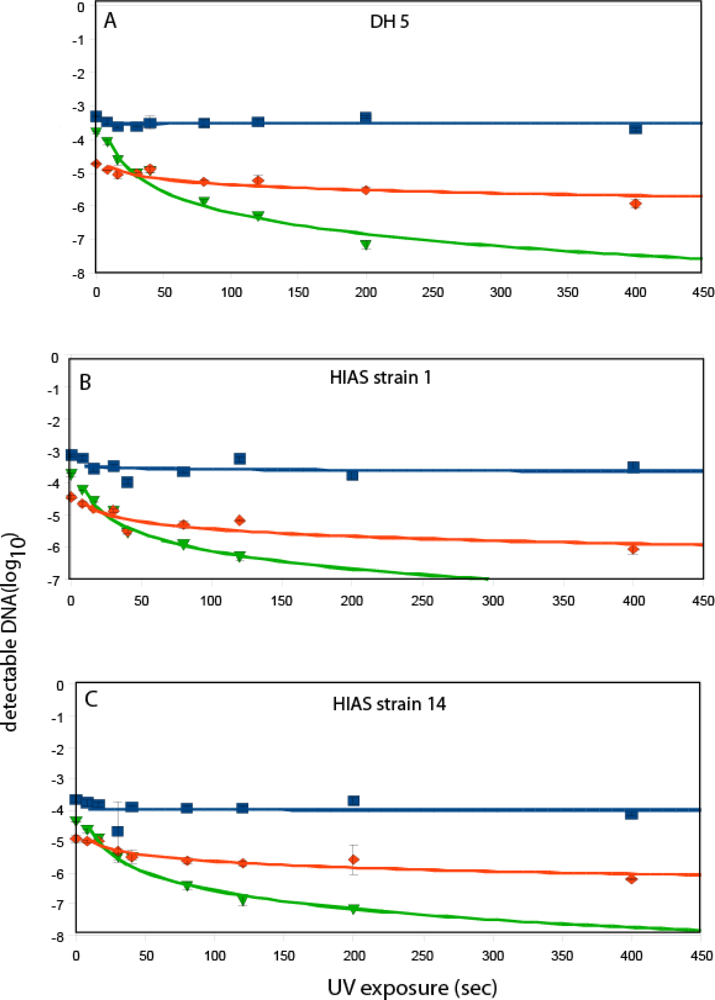
3. Results and Discussion
Acknowledgments
References and Notes
- Hijnen, WA; Beerendonk, EF; Medema, GJ. Inactivation credit of UV radiation for viruses, bacteria and protozoan (oo)cysts in water: A review. Water Res 2006, 40, 3–22. [Google Scholar]
- Sinha, RP; Hader, DP. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci 2002, 1, 225–236. [Google Scholar]
- Sarkar, G; Sommer, SS. Parameters affecting susceptibility of PCR contamination to UV inactivation. Bio. Techniques 1991, 10, 590–594. [Google Scholar]
- Swango, KL; Hudlow, WR; Timken, MD; Buoncristiani, MR. Developmental validation of a multiplex qPCR assay for assessing the quantity and quality of nuclear DNA in forensic samples. Forensic Sci. Int 2007, 170, 35–45. [Google Scholar]
- Niederstatter, H; Kochl, S; Grubwieser, P; Pavlic, M; Steinlechner, M; Parson, W. A modular real-time PCR concept for determining the quantity and quality of human nuclear and mitochondrial DNA. Forensic Sci. Int. Genet 2007, 1, 29–34. [Google Scholar]
- Trombert, A; Irazoqui, H; Martín, C; Zalazar, F. Evaluation of UV-C induced changes in Escherichia coli DNA using repetitive extragenic palindromic-polymerase chain reaction (REP-PCR). J. Photochem. Photobiol. B: Biol 2007, 89, 44–49. [Google Scholar]
- Hunter, PR. Drinking water and diarrhoeal disease due to Escherichia coli. J. Water Health 2003, 1, 65–72. [Google Scholar]
- Frahm, E; Obst, U. Application of the fluorogenic probe technique (TaqMan PCR) to the detection of Enterococcus spp. and Escherichia coli in water samples. J. Microbiol. Method 2003, 52, 123–131. [Google Scholar]
- Bustin, SA; Benes, V; Garson, JA; Hellemans, J; Huggett, J; Kubista, M; Mueller, R; Nolan, T; Pfaffl, MW; Shipley, GL; Vandesompele, J; Wittwer, CT. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 2009, 55, 611–622. [Google Scholar]
- Nadkarni, MA; Martin, FE; Jacques, NA; Hunter, N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 2002, 148, 257–266. [Google Scholar]


{kind=link}
| Sample # | Time (s) | UV dose (mWs/cm2) |
|---|---|---|
| 1 | 0 | not treated |
| 2 | 8 | 8 |
| 3 | 16 | 16 |
| 4 | 30 | 30 * |
| 5 | 40 | 40 ** |
| 6 | 80 | 80 |
| 7 | 120 | 120 |
| 8 | 200 | 200 |
| 9 | 400 | 400 |
| Amplicons | Thermocycling | Amplification | |||||
|---|---|---|---|---|---|---|---|
| Name | Primer sequence | Position * | Denaturation | Annealing | Synthesis | Efficiency | R2 |
| Short F | GAAGAAGCACCGGCTAAC | 529 | 95 °C – 30s | 50 °C – 30s | 72 °C – 30s | 0.43 | 1 |
| Short R | GCT TTACGCCCAGTCATTC | 611 | |||||
| Medium F | TCCTACGGGAGGCAGCAGT | 375 | 95 °C – 30s | 63 °C – 30s | 72 °C – 45s | 0.68 | 1 |
| Medium R | GGACTACCAGGGTATCTAATCCTGTT | 841 | |||||
| Long F | AAGAGTTTGATCATGGCTCA | 42 | 95 °C – 30s | 55° C – 30s | 72 °C – 90s | 0.54 | 0.98 |
| Long R | CGGTTACCTTGTTACGACTT | 1546 | |||||
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rudi, K.; Hagen, I.; Johnsrud, B.C.; Skjefstad, G.; Tryland, I. Different Length (DL) qPCR for Quantification of Cell Killing by UV-induced DNA Damage. Int. J. Environ. Res. Public Health 2010, 7, 3376-3381. https://doi.org/10.3390/ijerph7093376
Rudi K, Hagen I, Johnsrud BC, Skjefstad G, Tryland I. Different Length (DL) qPCR for Quantification of Cell Killing by UV-induced DNA Damage. International Journal of Environmental Research and Public Health. 2010; 7(9):3376-3381. https://doi.org/10.3390/ijerph7093376
Chicago/Turabian StyleRudi, Knut, Irina Hagen, Bente Carina Johnsrud, Guro Skjefstad, and Ingun Tryland. 2010. "Different Length (DL) qPCR for Quantification of Cell Killing by UV-induced DNA Damage" International Journal of Environmental Research and Public Health 7, no. 9: 3376-3381. https://doi.org/10.3390/ijerph7093376
APA StyleRudi, K., Hagen, I., Johnsrud, B. C., Skjefstad, G., & Tryland, I. (2010). Different Length (DL) qPCR for Quantification of Cell Killing by UV-induced DNA Damage. International Journal of Environmental Research and Public Health, 7(9), 3376-3381. https://doi.org/10.3390/ijerph7093376