Life-long Programming Implications of Exposure to Tobacco Smoking and Nicotine Before and Soon After Birth: Evidence for Altered Lung Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Effects of Maternal Tobacco Smoking on Lung Function and Respiratory Health of Offspring
3. Nicotine Uptake
4. Metabolism of Nicotine during Pregnancy
5. Nicotine and Oxidant/Antioxidant Status
6. Effects of Maternal Nicotine on Nutritional, Hormonal and Biochemical Profiles in the Offspring
7. Nicotine-Induced Body Malformations
8. Effect of Nicotine on the Development of the Lung
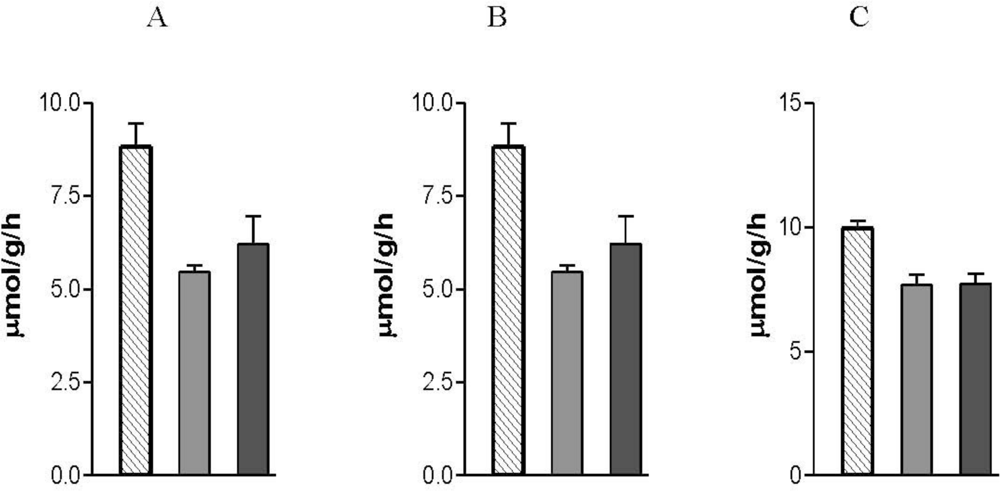
9. Effects of Nicotine on Metabolic Activity in the Lung
9.1. Energy Metabolism
9.2. Xenobiotic Metabolism
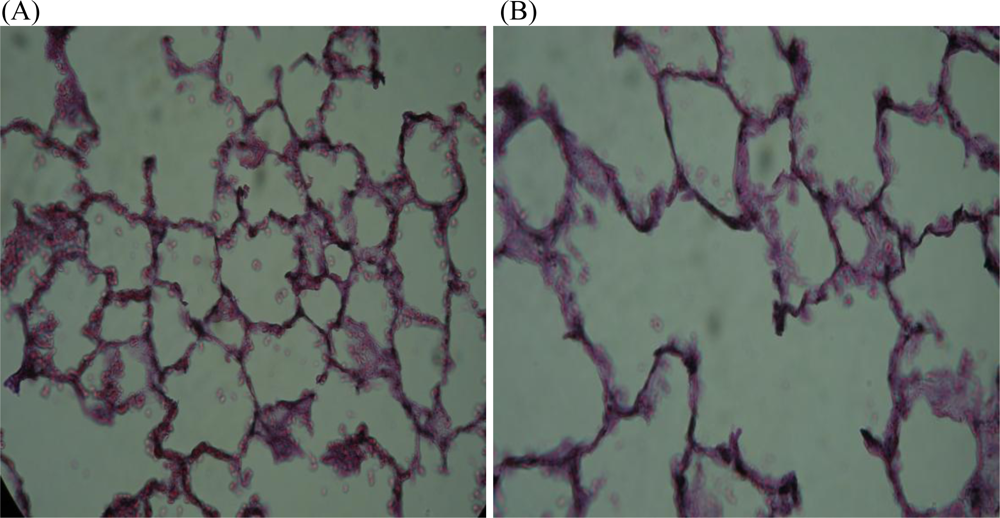
10. Effects of Nicotine on Structural Development of the Lungs
11. Nicotine and Cell Signaling: Apoptosis and Lung Development
12. Nicotine and Immune Response
13. Nicotine Replacement Therapy (NRT)
14. Conclusions
References
- Halima, BA; Sarra, K; Kais, R; Salwa, E; Najoua, G. Indicators of oxidative stress in weanling and pubertal rats following exposure to nicotine via milk. Hum. Exp. Toxicol 2010, 29, 489–496. [Google Scholar]
- Ozokutan, BH; Ozkan, KU; Sari, I; Inanc, F; Guldur, ME; Kilinc, M. Effects of maternal nicotine exposure during lactation on breast-fed rat pups. Biol. Neonate 2005, 88, 113–117. [Google Scholar]
- Oliveira, E; Pinheiro, CR; Santos-Silva, AP; Trevenzoli, IH; Abreu-Villaca, Y; Nogueira Neto, JF; Reis, AM; Passos, MC; Moura, EG; Lisboa, PC. Nicotine exposure affects mother’s and pup’s nutritional, biochemical, and hormonal profiles during lactation in rats. J. Endocrinol 2010, 205, 159–170. [Google Scholar]
- Jiménez Ruiz, CA. Tratamiento sustitutivo con nicotina en el embarazo. Arch. Bronconeumol 2006, 42, 404–409. [Google Scholar]
- Siu, EC; Tyndale, RF. Non-nicotinic therapies for smoking cessation. Annu. Rev. Pharmacol. Toxicol 2007, 47, 541–564. [Google Scholar]
- Gonzales, D; Rennard, SI; Nides, M; Oncken, C; Azoulay, S; Billing, CB; Watsky, EJ; Gong, J; Williams, KE; Reeves, KR. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs. sustained-release bupropion and placebo for smoking cessation: A randomized controlled trial. JAMA 2006, 296, 47–55. [Google Scholar]
- Potts, LA; Garwood, CL. Varenicline: The newest agent for smoking cessation. Am. J. Health Syst. Pharm 2007, 64, 1381–1384. [Google Scholar]
- Hofhuis, W; de Jongste, JC; Merkus, PJ. Adverse health effects of prenatal and postnatal tobacco smoke exposure on children. Arch. Dis. Child 2003, 88, 1086–1090. [Google Scholar]
- Lodrup Carlsen, KC; Jaakkola, JJ; Nafstad, P; Carlsen, KH. In utero exposure to cigarette smoking influences lung function at birth. Eur. Respir. J 1997, 10, 1774–1779. [Google Scholar]
- Stocks, J; Dezateux, C. The effect of parental smoking on lung function and development during infancy. Respirology 2003, 8, 266–285. [Google Scholar]
- Elliot, J; Carroll, N; Bosco, M; McCrohan, M; Robinson, P. Increased airway responsiveness and decreased alveolar attachment points following in utero smoke exposure in the guinea pig. Am. J. Respir. Crit. Care Med 2001, 163, 140–144. [Google Scholar]
- Landau, LI. Tobacco smoke exposure and tracking of lung function into adult life. Paediatr. Respir. Rev 2008, 9, 39–43. [Google Scholar]
- Gilliland, FD; Berhane, K; McConnell, R; Gauderman, WJ; Vora, H; Rappaport, EB; Avol, E; Peters, JM. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax 2000, 55, 271–276. [Google Scholar]
- Goksor, E; Amark, M; Alm, B; Gustafsson, PM; Wennergren, G. The impact of pre- and post-natal smoke exposure on future asthma and bronchial hyper-responsiveness. Acta Paediatr 2007, 96, 1030–1035. [Google Scholar]
- Henderson, AJ; Newson, RB; Rose-Zerilli, M; Ring, SM; Holloway, JW; Shaheen, SO. Maternal Nrf2 and gluthathione-S-transferase polymorphisms do not modify associations of prenatal tobacco smoke exposure with asthma and lung function in school-aged children. Thorax 2010, 65, 897–902. [Google Scholar]
- Upton, MN; Smith, GD; McConnachie, A; Hart, CL; Watt, GC. Maternal and personal cigarette smoking synergize to increase airflow limitation in adults. Am. J. Respir. Crit. Care Med 2004, 169, 479–487. [Google Scholar]
- Beyer, D; Mitfessel, H; Gillissen, A. Maternal smoking promotes chronic obstructive lung disease in the offspring as adults. Eur. J. Med. Res 2009, 14, 27–31. [Google Scholar]
- Brewer, BG; Roberts, AM; Rowell, PP. Short-term distribution of nicotine in the rat lung. Drug Alcohol Depend 2004, 75, 193–198. [Google Scholar]
- Matta, SG; Balfour, DJ; Benowitz, NL; Boyd, RT; Buccafusco, JJ; Caggiula, AR; Craig, CR; Collins, AC; Damaj, MI; Donny, EC; Gardiner, PS; Grady, SR; Heberlein, U; Leonard, SS; Levin, ED; Lukas, RJ; Markou, A; Marks, MJ; McCallum, SE; Parameswaran, N; Perkins, KA; Picciotto, MR; Quik, M; Rose, JE; Rothenfluh, A; Schafer, WR; Stolerman, IP; Tyndale, RF; Wehner, JM; Zirger, JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology 2007, 190, 269–319. [Google Scholar]
- Onuki, M; Yokoyama, K; Kimura, K; Sato, H; Nordin, RB; Naing, L; Morita, Y; Sakai, T; Kobayashi, Y; Araki, S. Assessment of urinary cotinine as a marker of nicotine absorption from tobacco leaves: A study on tobacco farmers in Malaysia. J. Occup. Health 2003, 45, 140–145. [Google Scholar]
- Sastry, BV; Chance, MB; Hemontolor, ME; Goddijn-Wessel, TA. Formation and retention of cotinine during placental transfer of nicotine in human placental cotyledon. Pharmacology 1998, 57, 104–116. [Google Scholar]
- Dahlstrom, A; Lundell, B; Curvall, M; Thapper, L. Nicotine and cotinine concentrations in the nursing mother and her infant. Acta Paediatr. Scand 1990, 79, 142–147. [Google Scholar]
- Luck, W; Nau, H; Hansen, R; Steldinger, R. Extent of nicotine and cotinine transfer to the human fetus, placenta and amniotic fluid of smoking mothers. Dev. Pharmacol. Ther 1985, 8, 384–395. [Google Scholar]
- Benowitz, NL; Jacob, P, III. Nicotine and carbon monoxide intake from high- and low-yield cigarettes. Clin. Pharmacol. Ther 1984, 36, 265–270. [Google Scholar]
- Hukkanen, J; Jacob, P, III; Benowitz, NL. Metabolism and disposition kinetics of nicotine. Pharmacol. Rev 2005, 57, 79–115. [Google Scholar]
- Henningfield, JE; Stapleton, JM; Benowitz, NL; Grayson, RF; London, ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend 1993, 33, 23–29. [Google Scholar]
- Suzuki, K; Horiguchi, T; Comas-Urrutia, AC; Mueller-Heubach, E; Morishima, HO; Adamsons, K. Placental transfer and distribution of nicotine in the pregnant rhesus monkey. Am. J. Obstet. Gynecol 1974, 119, 253–262. [Google Scholar]
- Dempsey, DA; Benowitz, NL. Risks and benefits of nicotine to aid smoking cessation in pregnancy. Drug Saf 2001, 24, 277–322. [Google Scholar]
- Frank, L; Sosenko, IR. Prenatal development of lung antioxidant enzymes in four species. J. Pediatr 1987, 110, 106–110. [Google Scholar]
- Hayashibe, H; Asayama, K; Dobashi, K; Kato, K. Prenatal development of antioxidant enzymes in rat lung, kidney, and heart: Marked increase in immunoreactive superoxide dismutases, glutathione peroxidase, and catalase in the kidney. Pediatr. Res 1990, 27, 472–475. [Google Scholar]
- Walther, FJ; Wade, AB; Warburton, D; Forman, HJ. Ontogeny of antioxidant enzymes in the fetal lamb lung. Exp. Lung Res 1991, 17, 39–45. [Google Scholar]
- Lambers, DS; Clark, KE. The maternal and fetal physiologic effects of nicotine. Semin. Perinatol 1996, 20, 115–126. [Google Scholar]
- Kleinsasser, NH; Sassen, AW; Semmler, MP; Harreus, UA; Licht, AK; Richter, E. The tobacco alkaloid nicotine demonstrates genotoxicity in human tonsillar tissue and lymphocytes. Toxicol. Sci 2005, 86, 309–317. [Google Scholar]
- Bruin, JE; Petre, MA; Raha, S; Morrison, KM; Gerstein, HC; Holloway, AC. Fetal and neonatal nicotine exposure in Wistar rats causes progressive pancreatic mitochondrial damage and beta cell dysfunction. PLoS One 2008, 3, e3371. [Google Scholar]
- Rehan, VK; Wang, Y; Sugano, S; Santos, J; Patel, S; Sakurai, R; Boros, LG; Lee, WP; Torday, JS. In utero nicotine exposure alters fetal rat lung alveolar type II cell proliferation, differentiation, and metabolism. Am. J. Physiol. Lung Cell Mol. Physiol 2007, 292, L323–L333. [Google Scholar]
- Guo, J; Chu, M; Abbeyquaye, T; Chen, CY. Persistent nicotine treatment potentiates amplification of the dihydrofolate reductase gene in rat lung epithelial cells as a consequence of Ras activation. J. Biol. Chem 2005, 280, 30422–30431. [Google Scholar]
- Hartwell, LH; Kastan, MB. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar]
- Noakes, PS; Thomas, R; Lane, C; Mori, TA; Barden, AE; Devadason, SG; Prescott, SL. Association of maternal smoking with increased infant oxidative stress at 3 months of age. Thorax 2007, 62, 714–717. [Google Scholar]
- Orhon, FS; Ulukol, B; Kahya, D; Cengiz, B; Baskan, S; Tezcan, S. The influence of maternal smoking on maternal and newborn oxidant and antioxidant status. Eur. J. Pediatr 2009, 168, 975–981. [Google Scholar]
- Husain, K; Scott, BR; Reddy, SK; Somani, SM. Chronic ethanol and nicotine interaction on rat tissue antioxidant defense system. Alcohol 2001, 25, 89–97. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Wallace, DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet 2005, 39, 359–407. [Google Scholar]
- Zaken, V; Kohen, R; Ornoy, A. Vitamins C and E improve rat embryonic antioxidant defense mechanism in diabetic culture medium. Teratology 2001, 64, 33–44. [Google Scholar]
- Oliveira, E; Moura, EG; Santos-Silva, AP; Fagundes, AT; Rios, AS; Abreu-Villaca, Y; Nogueira Neto, JF; Passos, MC; Lisboa, PC. Short- and long-term effects of maternal nicotine exposure during lactation on body adiposity, lipid profile, and thyroid function of rat offspring. J. Endocrinol 2009, 202, 397–405. [Google Scholar]
- Zhao, Z; Reece, EA. Nicotine-induced embryonic malformations mediated by apoptosis from increasing intracellular calcium and oxidative stress. Birth Defects Res. B Dev. Reprod. Toxicol 2005, 74, 383–391. [Google Scholar]
- Haustein, KO. Cigarette smoking, nicotine and pregnancy. Int. J. Clin. Pharmacol. Ther 1999, 37, 417–427. [Google Scholar]
- Ornoy, A. Embryonic oxidative stress as a mechanism of teratogenesis with special emphasis on diabetic embryopathy. Reprod. Toxicol 2007, 24, 31–41. [Google Scholar]
- Sekhon, HS; Jia, Y; Raab, R; Kuryatov, A; Pankow, JF; Whitsett, JA; Lindstrom, J; Spindel, ER. Prenatal nicotine increases pulmonary alpha7 nicotinic receptor expression and alters fetal lung development in monkeys. J. Clin. Invest 1999, 103, 637–647. [Google Scholar]
- Maneckjee, R; Minna, JD. Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ 1994, 5, 1033–1040. [Google Scholar]
- Maus, AD; Pereira, EF; Karachunski, PI; Horton, RM; Navaneetham, D; Macklin, K; Cortes, WS; Albuquerque, EX; Conti-Fine, BM. Human and rodent bronchial epithelial cells express functional nicotinic acetylcholine receptors. Mol. Pharmacol 1998, 54, 779–788. [Google Scholar]
- Pontieri, FE; Tanda, G; Orzi, F; Di Chiara, G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 1996, 382, 255–257. [Google Scholar]
- Wongtrakool, C; Roser-Page, S; Rivera, HN; Roman, J. Nicotine alters lung branching morphogenesis through the alpha7 nicotinic acetylcholine receptor. Am. J. Physiol. Lung Cell. Mol. Physiol 2007, 293, L611–L618. [Google Scholar]
- Sekhon, HS; Keller, JA; Benowitz, NL; Spindel, ER. Prenatal nicotine exposure alters pulmonary function in newborn rhesus monkeys. Am. J. Respir. Crit. Care Med 2001, 164, 989–994. [Google Scholar]
- Proskocil, BJ; Sekhon, HS; Jia, Y; Savchenko, V; Blakely, RD; Lindstrom, J; Spindel, ER. Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology 2004, 145, 2498–2506. [Google Scholar]
- Cattaneo, MG; D’Atri, F; Vicentini, LM. Mechanisms of mitogen-activated protein kinase activation by nicotine in small-cell lung carcinoma cells. Biochem. J 1997, 328, 499–503. [Google Scholar]
- Minna, JD. Nicotine exposure and bronchial epithelial cell nicotinic acetylcholine receptor expression in the pathogenesis of lung cancer. J. Clin. Invest 2003, 111, 31–33. [Google Scholar]
- Chen, CY; Liou, J; Forman, LW; Faller, DV. Differential regulation of discrete apoptotic pathways by Ras. J. Biol. Chem 1998, 273, 16700–16709. [Google Scholar]
- Heeschen, C; Jang, JJ; Weis, M; Pathak, A; Kaji, S; Hu, RS; Tsao, PS; Johnson, FL; Cooke, JP. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med 2001, 7, 833–839. [Google Scholar]
- Macklin, KD; Maus, AD; Pereira, EF; Albuquerque, EX; Conti-Fine, BM. Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther 1998, 287, 435–439. [Google Scholar]
- West, KA; Brognard, J; Clark, AS; Linnoila, IR; Yang, X; Swain, SM; Harris, C; Belinsky, S; Dennis, PA. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Invest 2003, 111, 81–90. [Google Scholar]
- Vogelstein, B; Kinzler, KW. p53 function and dysfunction. Cell 1992, 70, 523–526. [Google Scholar]
- O’Neil, JJ; Tierney, DF. Rat lung metabolism: glucose utilization by isolated perfused lungs and tissue slices. Am. J. Physiol 1974, 226, 867–873. [Google Scholar]
- Bourbon, J; Jost, A. Control of glycogen metabolism in the developing fetal lung. Pediatr. Res 1982, 16, 50–56. [Google Scholar]
- Gilden, C; Sevanian, A; Tierney, DF; Kaplan, SA; Barrett, CT. Regulation of fetal lung phosphatidyl choline synthesis by cortisol: Role of glycogen and glucose. Pediatr. Res 1977, 11, 845–848. [Google Scholar]
- Maniscalco, WM; Wilson, CM; Gross, I; Gobran, L; Rooney, SA; Warshaw, JB. Development of glycogen and phospholipid metabolism in fetal and newborn rat lung. Biochim. Biophys. Acta 1978, 530, 333–346. [Google Scholar]
- Salisbury-Murphy, S; Rubinstein, D; Beck, JC. Lipid metabolism in lung slices. Am. J. Physiol 1966, 211, 988–992. [Google Scholar]
- Post, M; Copland, I. Overview of lung development. Acta Pharmacol. Sin 2002, 23, S4–S7. [Google Scholar]
- Maritz, GS. Lung glycogen metabolism in suckling rats: A comparative study. Biol. Neonate 1988, 54, 100–106. [Google Scholar]
- Rhoades, RA. Net uptake of glucose, glycerol, and fatty acids by the isolated perfused rat lung. Am. J. Physiol 1974, 226, 144–149. [Google Scholar]
- Ito, T. Differentiation and proliferation of pulmonary neuroendocrine cells. Prog. Histochem. Cytochem 1999, 34, 247–322. [Google Scholar]
- Maritz, G. Pre- and postnatal carbohydrate metabolism of rat lung tissue. The effect of maternal nicotine exposure. Arch. Toxicol 1986, 59, 89–93. [Google Scholar]
- Ito, T; Noguchi, Y; Udaka, N; Kitamura, H; Satoh, S. Glucose transporter expression in developing fetal lungs and lung neoplasms. Histol. Histopathol 1999, 14, 895–904. [Google Scholar]
- Iba, MM; Fung, J; Pak, YW; Thomas, PE; Fisher, H; Sekowski, A; Halladay, AK; Wagner, GC. Dose-dependent up-regulation of rat pulmonary, renal, and hepatic cytochrome P-450 (CYP) 1A expression by nicotine feeding. Drug Metab. Dispos 1999, 27, 977–982. [Google Scholar]
- Maritz, GS. Maternal nicotine exposure and carbohydrate metabolism of fetal and neonatal lung tissue: response to nicotine withdrawal. Respiration 1987, 51, 232–240. [Google Scholar]
- Maritz, GS; Burger, B. The influence of maternal nicotine exposure on neonatal lung carbohydrate metabolism. Cell Biol. Int. Rep 1992, 16, 1229–1236. [Google Scholar]
- Kondoh, H; Lleonart, ME; Gil, J; Wang, J; Degan, P; Peters, G; Martinez, D; Carnero, A; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res 2005, 65, 177–185. [Google Scholar]
- Zwerschke, W; Mazurek, S; Stockl, P; Hutter, E; Eigenbrodt, E; Jansen-Durr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J 2003, 376, 403–411. [Google Scholar]
- Kondoh, H; Lleonart, ME; Bernard, D; Gil, J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol. Histopathol 2007, 22, 85–90. [Google Scholar]
- Hukkanen, J; Pelkonen, O; Raunio, H. Expression of xenobiotic-metabolizing enzymes in human pulmonary tissue: possible role in susceptibility for ILD. Eur. Respir. J. Suppl 2001, 32, 122s–126s. [Google Scholar]
- Russell, MA; Feyerabend, C. Cigarette smoking: A dependence on high-nicotine boli. Drug Metab. Rev 1978, 8, 29–57. [Google Scholar]
- Lee, CZ; Royce, FH; Denison, MS; Pinkerton, KE. Effect of in utero and postnatal exposure to environmental tobacco smoke on the developmental expression of pulmonary cytochrome P450 monooxygenases. J. Biochem. Mol. Toxicol 2000, 14, 121–130. [Google Scholar]
- Luck, W; Nau, H. Nicotine and cotinine concentrations in serum and milk of nursing smokers. Br. J. Clin. Pharmacol 1984, 18, 9–15. [Google Scholar]
- Raunio, H; Hakkola, J; Hukkanen, J; Lassila, A; Paivarinta, K; Pelkonen, O; Anttila, S; Piipari, R; Boobis, A; Edwards, RJ. Expression of xenobiotic-metabolizing CYPs in human pulmonary tissue. Exp. Toxicol. Pathol 1999, 51, 412–417. [Google Scholar]
- Gamieldien, K; Maritz, GS. Postnatal expression of cytochrome P450 1A1, 2A3, and 2B1 mRNA in neonatal rat lung: Influence of maternal nicotine exposure. Exp. Lung Res 2004, 30, 121–133. [Google Scholar]
- Carmella, SG; Borukhova, A; Akerkar, SA; Hecht, SS. Analysis of human urine for pyridine-N-oxide metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific lung carcinogen. Cancer Epidemiol. Biomarkers Prev 1997, 6, 113–120. [Google Scholar]
- Kauffman, SL; Burri, PH; Weibel, ER. The postnatal growth of the rat lung. II. Autoradiography. Anat. Rec 1974, 180, 63–76. [Google Scholar]
- Massaro, GD; Massaro, D. Formation of pulmonary alveoli and gas-exchange surface area: Quantitation and regulation. Annu. Rev. Physiol 1996, 58, 73–92. [Google Scholar]
- Coalson, JJ; Winter, V; deLemos, RA. Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med 1995, 152, 640–646. [Google Scholar]
- Margraf, LR; Tomashefski, JF, Jr; Bruce, MC; Dahms, BB. Morphometric analysis of the lung in bronchopulmonary dysplasia. Am. Rev. Respir. Dis 1991, 143, 391–400. [Google Scholar]
- Kida, K; Thurlbeck, WM. The effects of beta-aminopropionitrile on the growing rat lung. Am. J. Pathol 1980, 101, 693–710. [Google Scholar]
- Noguchi, A; Samaha, H. Developmental changes in tropoelastin gene expression in the rat lung studied by in situ hybridization. Am. J. Respir. Cell. Mol. Biol 1991, 5, 571–578. [Google Scholar]
- Nakamura, Y; Romberger, DJ; Tate, L; Ertl, RF; Kawamoto, M; Adachi, Y; Mio, T; Sisson, JH; Spurzem, JR; Rennard, SI. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am. J. Respir. Crit. Care Med 1995, 151, 1497–1503. [Google Scholar]
- Elliot, JG; Carroll, NG; James, AL; Robinson, PJ. Airway alveolar attachment points and exposure to cigarette smoke in utero. Am. J. Respir. Crit. Care Med 2003, 167, 45–49. [Google Scholar]
- Holz, O; Zuhlke, I; Jaksztat, E; Muller, KC; Welker, L; Nakashima, M; Diemel, KD; Branscheid, D; Magnussen, H; Jorres, RA. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur. Respir. J 2004, 24, 575–579. [Google Scholar]
- Muller, KC; Welker, L; Paasch, K; Feindt, B; Erpenbeck, VJ; Hohlfeld, JM; Krug, N; Nakashima, M; Branscheid, D; Magnussen, H; Jorres, RA; Holz, O. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir. Res 2006, 7, 32. [Google Scholar]
- Verbeken, EK; Cauberghs, M; Mertens, I; Clement, J; Lauweryns, JM; van de Woestijne, KP. The senile lung. Comparison with normal and emphysematous lungs. 1. Structural aspects. Chest 1992, 101, 793–799. [Google Scholar]
- Naimark, A. Nonventilatory functions of the lung. Summary. Am. Rev. Respir. Dis 1977, 115, 93–98. [Google Scholar]
- Massaro, GD; Gail, DB; Massaro, D. Lung oxygen consumption and mitochondria of alveolar epithelial and endothelial cells. J. Appl. Physiol 1975, 38, 588–592. [Google Scholar]
- Paul, RJ. Functional compartmentalization of oxidative and glycolytic metabolism in vascular smooth muscle. Am. J. Physiol 1983, 244, C399–409. [Google Scholar]
- Contran, RS; Kumar, V; Robbins, SL. Cellular injury and adaptation. In Pathologic Basis of Disease, 4th ed; Robbins, SL, Ed.; WB Saunders Co: Philadelphia, PA, USA, 1989; pp. 16–38. [Google Scholar]
- Witsch, IH. Proliferation of type II alveolar cells: A review of common responses in toxic lung injury. Toxicology 1976, 5, 267–277. [Google Scholar]
- Maritz, GS; Thomas, RA. The influence of maternal nicotine exposure on the interalveolar septal status of neonatal rat lung. Cell Biol. Int 1994, 18, 747–757. [Google Scholar]
- Berthiaume, Y; Voisin, G; Dagenais, A. The alveolar type I cells: The new knight of the alveolus? J. Physiol 2006, 572, 609–610. [Google Scholar]
- Maritz, GS; Thomas, RA. Maternal nicotine exposure: response of type II pneumocytes of neonatal rat pups. Cell Biol. Int 1995, 19, 323–331. [Google Scholar]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res 2001, 2, 33–46. [Google Scholar]
- Hodes, RJ. Telomere length, aging, and somatic cell turnover. J. Exp. Med 1999, 190, 153–156. [Google Scholar]
- Rehan, VK; Asotra, K; Torday, JS. The effects of smoking on the developing lung: Insights from a biologic model for lung development, homeostasis, and repair. Lung 2009, 187, 281–289. [Google Scholar]
- Maritz, G. Maternal nicotine exposure induces microscopic emphysema in neonatal rat lung. Pathophysiology 1997, 4, 1–7. [Google Scholar]
- Maritz, GS; Windvogel, S. Is maternal copper supplementation during alveolarization protecting the developing rat lung against the adverse effects of maternal nicotine exposure? A morphometric study. Exp. Lung. Res 2003, 29, 243–260. [Google Scholar]
- Collins, MH; Moessinger, AC; Kleinerman, J; Bassi, J; Rosso, P; Collins, AM; James, LS; Blanc, WA. Fetal lung hypoplasia associated with maternal smoking: A morphometric analysis. Pediatr. Res 1985, 19, 408–412. [Google Scholar]
- Tsuji, T; Aoshiba, K; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell. Mol. Biol 2004, 31, 643–649. [Google Scholar]
- Maritz, GS; Windvogel, S. Chronic maternal nicotine exposure during gestation and lactation and the development of the lung parenchyma in the offspring. Response to nicotine withdrawal. Pathophysiology 2003, 10, 69–75. [Google Scholar]
- Balakrishnan, A; Menon, VP. Role of hesperidin on nicotine toxicity. Int. J. Pharmacol 2006, 2, 664–669. [Google Scholar]
- Maritz, G; Windvogel, S. Does maternal nicotine exposure during different phases of lung development influence the program that regulates the maintenance of lung integrity in the offspring? A comparative morphologic and morphometric study. Trends Comp. Biochem. Physiol 2005, 11, 69–75. [Google Scholar]
- Kalpana, C; Menon, VP. Modulatory effects of curcumin on lipid peroxidation and antioxidant status during nicotine-induced toxicity. Pol. J. Pharmacol 2004, 56, 581–586. [Google Scholar]
- Soma, T; Kaganoi, J; Kawabe, A; Kondo, K; Imamura, M; Shimada, Y. Nicotine induces the fragile histidine triad methylation in human esophageal squamous epithelial cells. Int. J. Cancer 2006, 119, 1023–1027. [Google Scholar]
- Holloway, AC; Cuu, DQ; Morrison, KM; Gerstein, HC; Tarnopolsky, MA. Transgenerational effects of fetal and neonatal exposure to nicotine. Endocrine 2007, 31, 254–259. [Google Scholar]
- White, E. Life, death, and the pursuit of apoptosis. Genes Dev 1996, 10, 1–15. [Google Scholar]
- Wertz, IE; Hanley, MR. Diverse molecular provocation of programmed cell death. Trends Biochem. Sci 1996, 21, 359–364. [Google Scholar]
- Schittny, JC; Djonov, V; Fine, A; Burri, PH. Programmed cell death contributes to postnatal lung development. Am. J. Respir. Cell. Mol. Biol 1998, 18, 786–793. [Google Scholar]
- Bruce, MC; Honaker, CE; Cross, RJ. Lung fibroblasts undergo apoptosis following alveolarization. Am. J. Respir. Cell Mol. Biol 1999, 20, 228–236. [Google Scholar]
- Wright, SC; Zhong, J; Zheng, H; Larrick, JW. Nicotine inhibition of apoptosis suggests a role in tumor promotion. FASEB J 1993, 7, 1045–1051. [Google Scholar]
- Fischer, S; Spiegelhalder, B; Eisenbarth, J; Preussmann, R. Investigations on the Origin of Tobacco-Specific Nitrosamines in Mainstream Smoke of Cigarettes. Carcinogenesis 1990, 11, 723–730. [Google Scholar]
- Hecht, SS; Hoffmann, D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis 1988, 9, 875–884. [Google Scholar]
- Heusch, WL; Maneckjee, R. Signalling pathways involved in nicotine regulation of apoptosis of human lung cancer cells. Carcinogenesis 1998, 19, 551–556. [Google Scholar]
- Maritz, GS; Matthews, HL; Aalbers, J. Maternal copper supplementation protects the neonatal rat lung against the adverse effects of maternal nicotine exposure. Reprod. Fertil. Dev 2000, 12, 97–103. [Google Scholar]
- Schuller, HM; Jull, BA; Sheppard, BJ; Plummer, HK. Interaction of tobacco-specific toxicants with the neuronal alpha(7) nicotinic acetylcholine receptor and its associated mitogenic signal transduction pathway: potential role in lung carcinogenesis and pediatric lung disorders. Eur. J. Pharmacol 2000, 393, 265–277. [Google Scholar]
- Brooks, DR; Mucci, LA; Hatch, EE; Cnattingius, S. Maternal smoking during pregnancy and risk of brain tumors in the offspring. A prospective study of 1.4 million Swedish births. Cancer Cause. Control 2004, 15, 997–1005. [Google Scholar]
- Ng, SP; Zelikoff, JT. Smoking during pregnancy: subsequent effects on offspring immune competence and disease vulnerability in later life. Reprod. Toxicol 2007, 23, 428–437. [Google Scholar]
- Argentin, G; Cicchetti, R. Genotoxic and antiapoptotic effect of nicotine on human gingival fibroblasts. Toxicol. Sci 2004, 79, 75–81. [Google Scholar]
- Dietert, R. Distinguishing environmental causes of immune dysfunction from pediatric triggers of disease. Open Pediat. Med. J 2009, 3, 38–44. [Google Scholar]
- Mishra, NC; Rir-sima-ah, J; Langley, RJ; Singh, SP; Pena-Philippides, JC; Koga, T; Razani-Boroujerdi, S; Hutt, J; Campen, M; Kim, KC; Tesfaigzi, Y; Sopori, ML. Nicotine primarily suppresses lung Th2 but not goblet cell and muscle cell responses to allergens. J. Immunol 2008, 180, 7655–7663. [Google Scholar]
- Benowitz, N; Dempsey, D. Pharmacotherapy for smoking cessation during pregnancy. Nicotine Tob. Res 2004, 6, S189–S202. [Google Scholar]
- Balakrishnan, A; Menon, VP. Antioxidant properties of hesperidin in nicotine-induced lung toxicity. Fundam. Clin. Pharmacol 2007, 21, 535–546. [Google Scholar]





© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maritz, G.S.; Harding, R. Life-long Programming Implications of Exposure to Tobacco Smoking and Nicotine Before and Soon After Birth: Evidence for Altered Lung Development. Int. J. Environ. Res. Public Health 2011, 8, 875-898. https://doi.org/10.3390/ijerph8030875
Maritz GS, Harding R. Life-long Programming Implications of Exposure to Tobacco Smoking and Nicotine Before and Soon After Birth: Evidence for Altered Lung Development. International Journal of Environmental Research and Public Health. 2011; 8(3):875-898. https://doi.org/10.3390/ijerph8030875
Chicago/Turabian StyleMaritz, Gert S., and Richard Harding. 2011. "Life-long Programming Implications of Exposure to Tobacco Smoking and Nicotine Before and Soon After Birth: Evidence for Altered Lung Development" International Journal of Environmental Research and Public Health 8, no. 3: 875-898. https://doi.org/10.3390/ijerph8030875
APA StyleMaritz, G. S., & Harding, R. (2011). Life-long Programming Implications of Exposure to Tobacco Smoking and Nicotine Before and Soon After Birth: Evidence for Altered Lung Development. International Journal of Environmental Research and Public Health, 8(3), 875-898. https://doi.org/10.3390/ijerph8030875