Concurrent Waldenstrom’s Macroglobulinemia and Myelodysplastic Syndrome with a Sequent t(10;13)(p13;q22) Translocation


 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
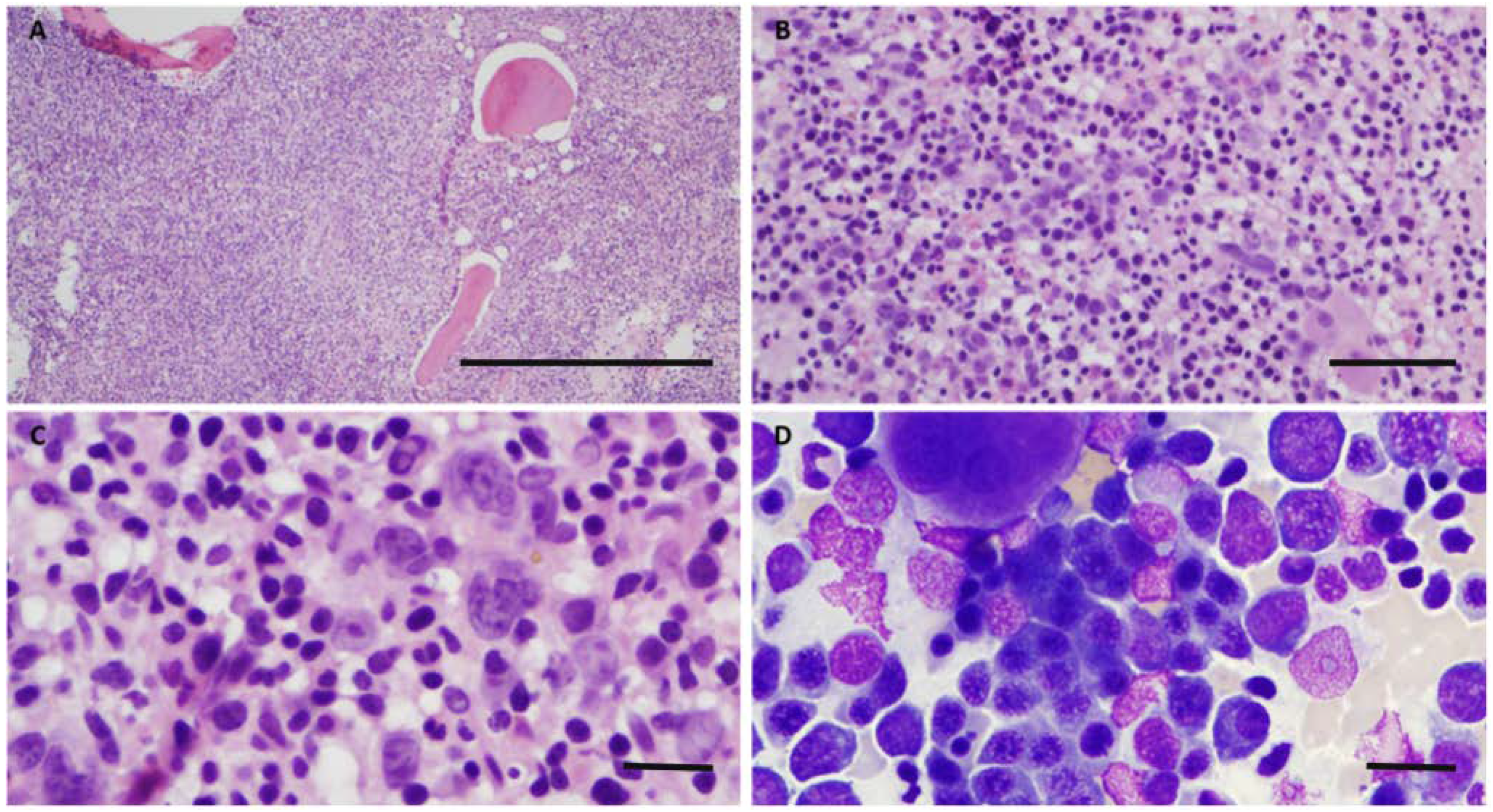
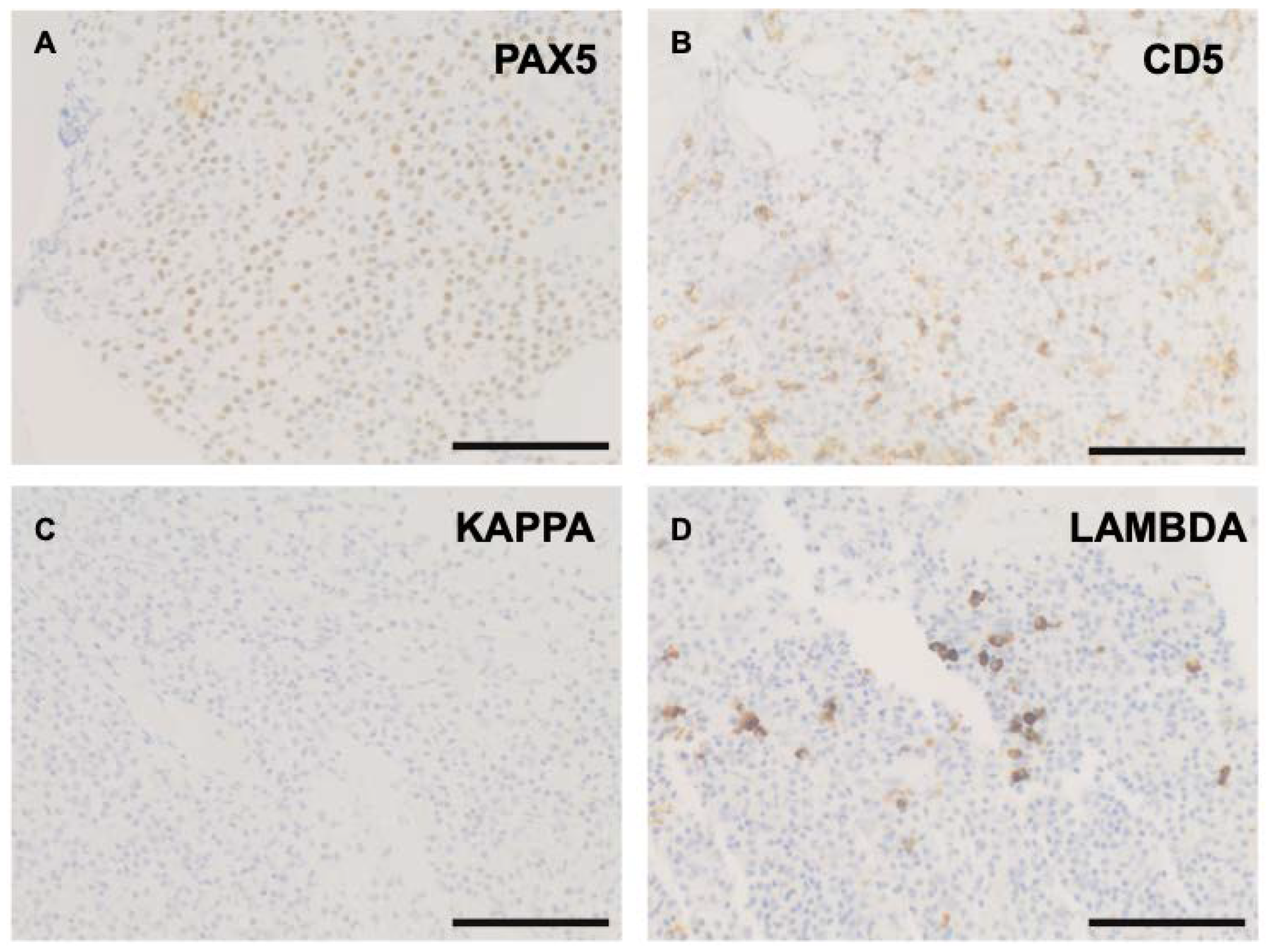
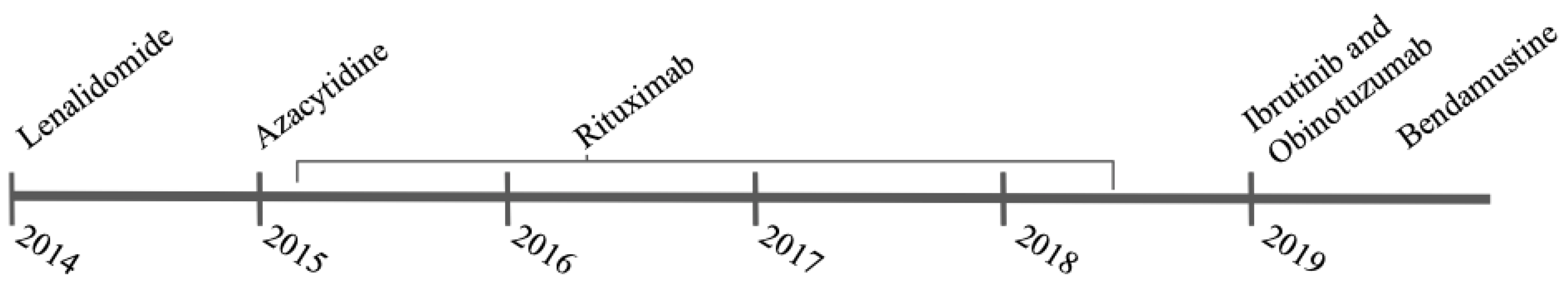
2. Case
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tefferi, A.; Vardiman, J.W. Myelodysplastic syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; De Santis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Treon, S.P.; Al-Katib, A.; Fonseca, R.; Greipp, P.R.; McMaster, M.L. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Blann, M.M.; Velagaleti, G.V.; Morgan, D.L.; Martinez, R.E.; Conlin, P.A.; Tonk, V.S. Interstitial deletion of 20q in a patient with Waldenstrom macroglobulinemia following chemotherapy. Cancer Genet Cytogenet. 2002, 132, 145–148. [Google Scholar] [CrossRef]
- Liu, Y.C.; Miyazawa, K.; Sashida, G.; Kodama, A.; Ohyashiki, K. Deletion (20q) as the sole abnormality in Waldenstrom macroglobulinemia suggests distinct pathogenesis of 20q11 anomaly. Cancer Genet Cytogenet. 2006, 169, 69–72. [Google Scholar] [CrossRef]
- Kitahara, T.; Umezu, T.; Ando, K.; Kodama, A.; Ohyashiki, J.H.; Ohyashiki, K. Non-random chromosomal deletion clustering at 20q in Waldenstrom macroglobulinemia. Hematology 2011, 16, 139–142. [Google Scholar] [CrossRef]
- Mori, M.; Terui, Y.; Ikeda, M.; Tomizuka, H.; Uwai, M.; Kasahara, T.; Kubota, N.; Itoh, T.; Mishima, Y.; Douzono-Tanaka, M.; et al. Beta(2)-microglobulin identified as an apoptosis-inducing factor and its characterization. Blood 1999, 94, 2744–2753. [Google Scholar]
- Duan, S.; Cermak, L.; Pagan, J.K.; Rossi, M.; Martinengo, C.; di Celle, P.F.; Chapuy, B.; Shipp, M.; Chiarle, R.; Pagano, M. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 2012, 481, 90–93. [Google Scholar] [CrossRef] [Green Version]
- Kanasugi, J.; Hanamura, I.; Ota, A.; Karnan, S.; Lam, V.Q.; Mizuno, S.; Wahiduzzaman, M.; Rahman, M.L.; Hyodo, T.; Konishi, H.; et al. Biallelic loss of FAM46C triggers tumor growth with concomitant activation of Akt signaling in multiple myeloma cells. Cancer Sci. 2020, 111, 1663–1675. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Benson, J.T.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Melton, L.J.; Rajkumar, S.V. Progression in smoldering Waldenstrom macroglobulinemia: Long-term results. Blood 2012, 119, 4462–4466. [Google Scholar] [CrossRef] [Green Version]
- McMaster, M.L. Familial Waldenstrom’s macroglobulinemia. Semin. Oncol. 2003, 30, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, K.; Kishikawa, Y.; Satoh, C.; Tamura, H.; Dan, K.; Hayashi, A. Diagnostic application of flow cytometric characteristics of CD34+ cells in low-grade myelodysplastic syndromes. Blood 2006, 108, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Montes, M.C.; Garcia-Sanz, R.; Ocio, E.M.; Alonso, J.; de Las Heras, N.; Escalante, F.; Cuello, R.; de Coca, A.G.; Galende, J.; et al. Multiparameter flow cytometry for the identification of the Waldenstrom’s clone in IgM-MGUS and Waldenstrom’s Macroglobulinemia: New criteria for differential diagnosis and risk stratification. Leukemia 2014, 28, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; World Health Organization; International Agency for Research on Cancer. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017.
- Treon, S.P.; Hunter, Z.R.; Aggarwal, A.; Ewen, E.P.; Masota, S.; Lee, C.; Santos, D.D.; Hatjiharissi, E.; Xu, L.; Leleu, X.; et al. Characterization of familial Waldenstrom’s macroglobulinemia. Ann. Oncol. 2006, 17, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Anagnostopoulos, A.; Anderson, K.; Branagan, A.R.; Coleman, M.; Frankel, S.R.; Giralt, S.; Levine, T.; Munshi, N.; Pestronk, A.; et al. Treatment recommendations in Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, H.; Young, K.H. The PTEN tumor suppressor gene and its role in lymphoma pathogenesis. Aging 2015, 7, 1032–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cao, X.; Sun, R.; Tang, C.; Tzankov, A.; Zhang, J.; Manyam, G.C.; Xiao, M.; Miao, Y.; Jabbar, K.; et al. Clinical Significance of PTEN Deletion, Mutation, and Loss of PTEN Expression in De Novo Diffuse Large B-Cell Lymphoma. Neoplasia 2018, 20, 574–593. [Google Scholar] [CrossRef] [PubMed]
- Post, K.L.; Belmadani, M.; Ganguly, P.; Meili, F.; Dingwall, R.; McDiarmid, T.A.; Meyers, W.M.; Herrington, C.; Young, B.P.; Callaghan, D.B.; et al. Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat. Commun. 2020, 11, 2073. [Google Scholar] [CrossRef]
- Liu, Y.F.; Wang, B.Y.; Zhang, W.N.; Huang, J.Y.; Li, B.S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hermanson, M.; Grander, D.; Merup, M.; Wu, X.; Heyman, M.; Rasool, O.; Juliusson, G.; Gahrton, G.; Detlofsson, R.; et al. 13q Deletions in Lymphoid Malignancies. Blood 1995, 86, 1911–1915. [Google Scholar] [CrossRef]
- Coignet, L.J.; Lima, C.S.; Min, T.; Streubel, B.; Swansbury, J.; Telford, N.; Swanton, S.; Bowen, A.; Nagai, M.; Catovsky, D.; et al. Myeloid- and lymphoid-specific breakpoint cluster regions in chromosome band 13q14 in acute leukemia. Genes Chromosomes Cancer 1999, 25, 222–229. [Google Scholar] [CrossRef]
- Joy, H.P. Chromosomal and genetic abnormalities in myeloma. Clin. Lab. Haematol. 2002, 24, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeRosa, P.A.; Roche, K.C.; Nava, V.E.; Singh, S.; Liu, M.-L.; Agarwal, A. Concurrent Waldenstrom’s Macroglobulinemia and Myelodysplastic Syndrome with a Sequent t(10;13)(p13;q22) Translocation. Curr. Oncol. 2022, 29, 4587-4592. https://doi.org/10.3390/curroncol29070363
DeRosa PA, Roche KC, Nava VE, Singh S, Liu M-L, Agarwal A. Concurrent Waldenstrom’s Macroglobulinemia and Myelodysplastic Syndrome with a Sequent t(10;13)(p13;q22) Translocation. Current Oncology. 2022; 29(7):4587-4592. https://doi.org/10.3390/curroncol29070363
Chicago/Turabian StyleDeRosa, Peter A., Kyle C. Roche, Victor E. Nava, Sunita Singh, Min-Ling Liu, and Anita Agarwal. 2022. "Concurrent Waldenstrom’s Macroglobulinemia and Myelodysplastic Syndrome with a Sequent t(10;13)(p13;q22) Translocation" Current Oncology 29, no. 7: 4587-4592. https://doi.org/10.3390/curroncol29070363
APA StyleDeRosa, P. A., Roche, K. C., Nava, V. E., Singh, S., Liu, M. -L., & Agarwal, A. (2022). Concurrent Waldenstrom’s Macroglobulinemia and Myelodysplastic Syndrome with a Sequent t(10;13)(p13;q22) Translocation. Current Oncology, 29(7), 4587-4592. https://doi.org/10.3390/curroncol29070363