
DFT Study of Heteronuclear (TMFeO3)x Molecular Clusters (Where TM = Sc, Ti, Fe and x = 2, 4, 8) for Photocatalytic and Photovoltaic Applications


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
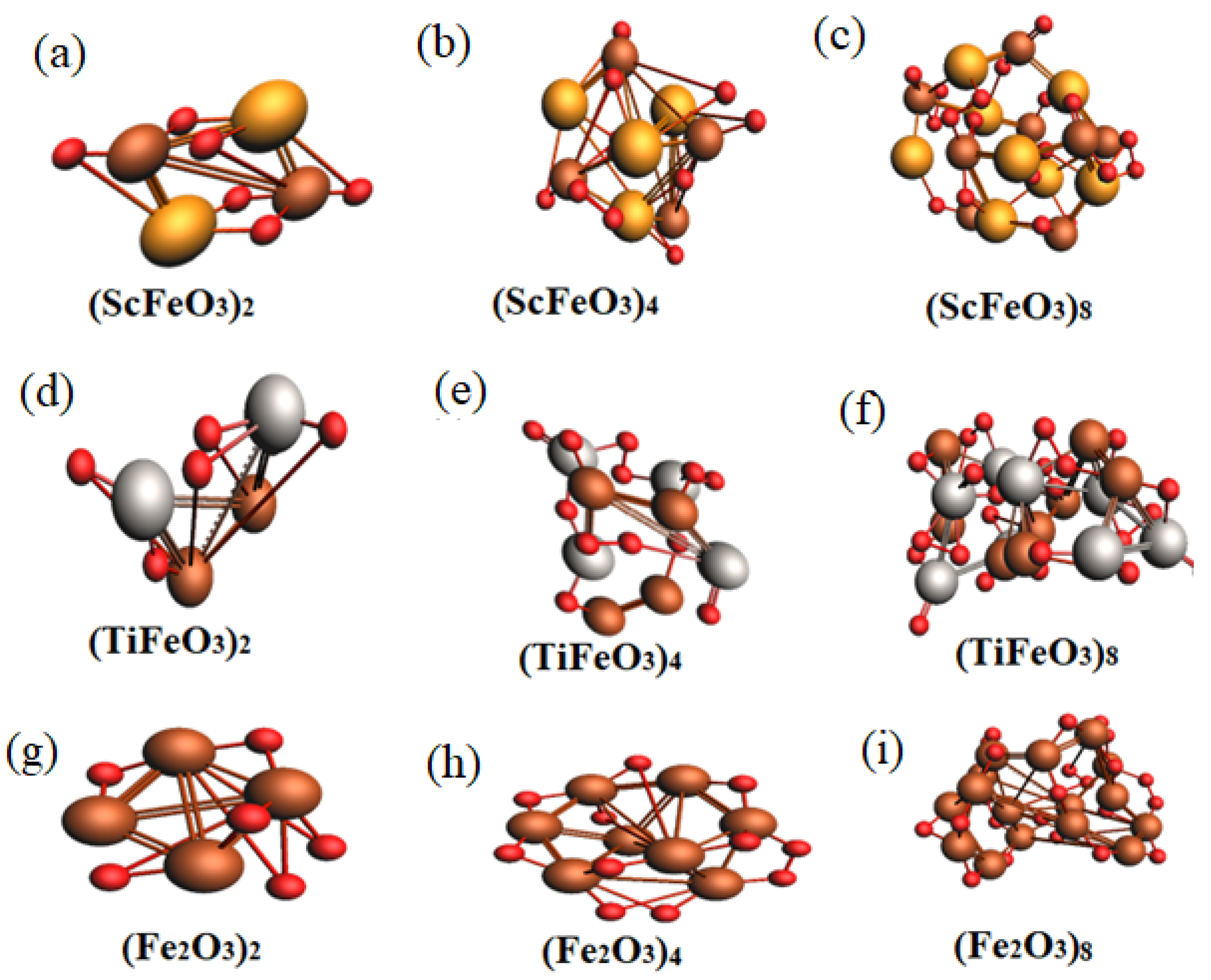
3.1. Structural Properties of x Molecular Clusters
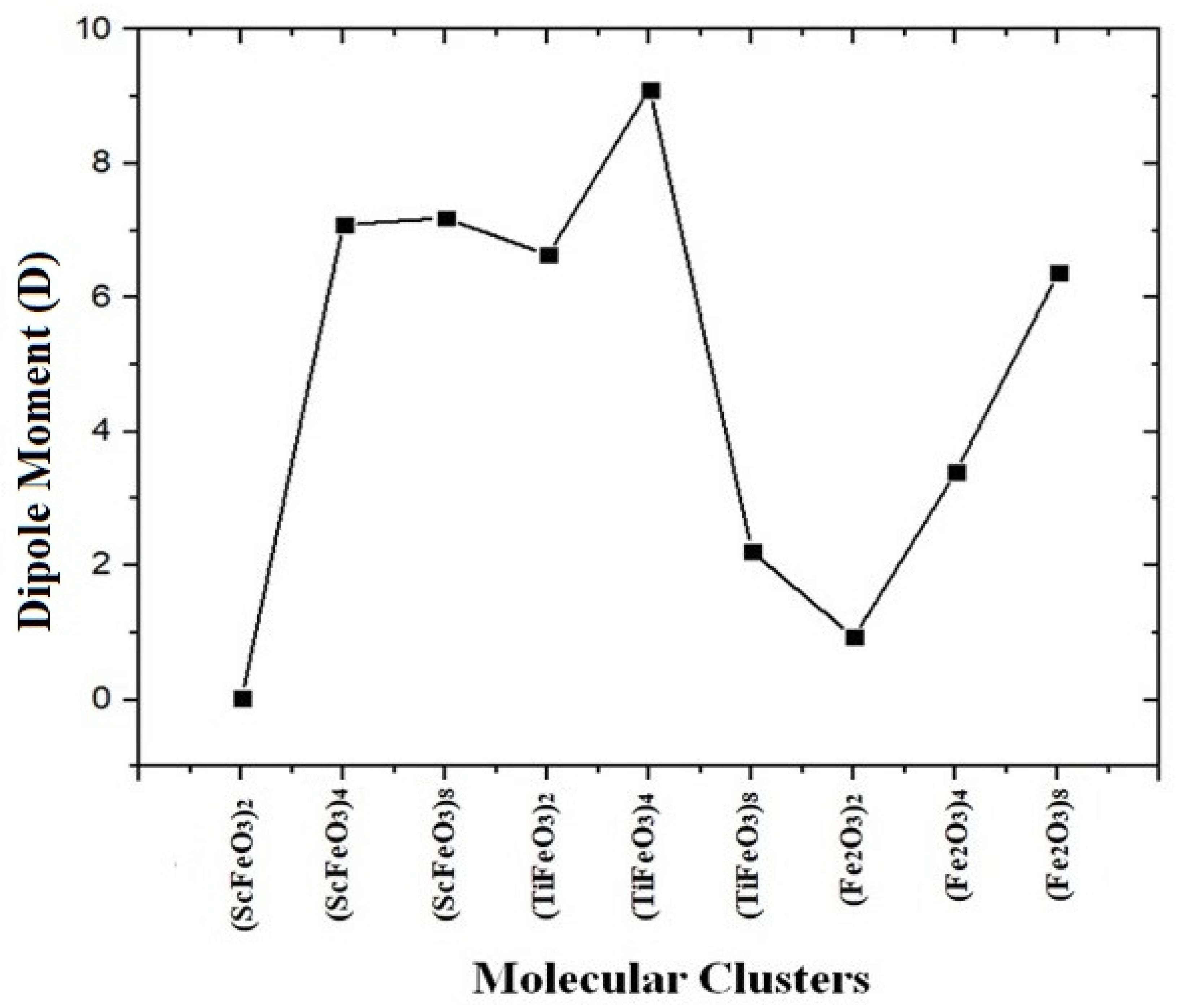
3.2. Electronic Properties of Molecular Clusters
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liu, W.-J.; Jiang, H.; Yu, H.-Q. Development of Biochar-Based Functional Materials: Toward a Sustainable Platform Carbon Material. Chem. Rev. 2015, 115, 12251–12285. [Google Scholar] [CrossRef]
- Ibrahim, M.E.; Eldin, E.T.; Elzoghby, S.F.; Izzularab, M.A.; Abd-Elhady, A.M. The Role of the Accumulated Surface Charge on Nanoparticles in Improving the Breakdown Strength of Liquid and Solid Insulation. Energies 2022, 15, 4860. [Google Scholar] [CrossRef]
- Guedri, K.; Raizah, Z.; Tag-Eldin, E.; Ashraf, W.; Khan, U.; Galal, A.M. Thermal efficiency in hybrid (Al2O3-CuO/H2O) and tri-hybrid (Al2O3-CuO-Cu/H2O) nanofluids between converging/diverging channel with viscous dissipation function: Numerical analysis. Front. Chem. 2022, 10, 960369. [Google Scholar]
- Lu, Y.; Chen, W. Sub-nanometre sized metal clusters: From synthetic challenges to the unique property discoveries. Chem. Soc. Rev. 2012, 41, 3594–3623. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liu, S.; Yi, Y.; Rong, H.; Zhang, J. Catalytic nanomaterials toward atomic levels for biomedical applications: From metal clusters to single-atom catalysts. ACS Nano 2021, 15, 2005–2037. [Google Scholar] [CrossRef]
- Abbas, Q.; Siyal, S.H.; Mateen, A.; Hassan, N.U.; Idrees, A.; Rehman, Z.U.; El Din, E.M.T.; Bajaber, M.A.; Javed, M.S. Hydrothermal Synthesis of Binder-Free Metallic NiCo2O4 Nano-Needles Supported on Carbon Cloth as an Advanced Electrode for Supercapacitor Applications. Materials 2022, 15, 4499. [Google Scholar] [CrossRef]
- Zhao, X.; Pei, G.; Xu, S.; Kong, C.; Yang, Z.; Yang, T. Endohedral group-14-element clusters TM@E9 (TM = Co, Ni, Cu; E = Ge, Sn, Pb) and their low-dimensional nanostructures: A first-principles study. Phys. Chem. Chem. Phys. 2021, 23, 20654–20665. [Google Scholar] [CrossRef]
- Kawasaki, N.; Wang, H.; Nakanishi, R.; Hamanaka, S.; Kitaura, R.; Shinohara, H.; Yokoyama, T.; Yoshikawa, H.; Awaga, K. Nanohybridization of Polyoxometalate Clusters and Single-Wall Carbon Nanotubes: Applications in Molecular Cluster Batteries. Angew. Chem. 2011, 123, 3533–3536. [Google Scholar] [CrossRef]
- Hang, X.; Bi, Y. Thiacalix [4] arene-supported molecular clusters for catalytic applications. Dalton Trans. 2021, 50, 3749–3758. [Google Scholar] [CrossRef]
- Sgibnev, Y.; Cattaruzza, E.; Dubrovin, V.; Vasilev, V.; Nikonorov, N. Photo-Thermo-Refractive Glasses Doped with Silver Molecular Clusters as Luminescence Downshifting Material for Photovoltaic Applications. Part. Part. Syst. Charact. 2018, 35. [Google Scholar] [CrossRef]
- Alberi, K.; Nardelli, M.B.; Zakutayev, A.; Mitas, L.; Curtarolo, S.; Jain, A.; Perkins, J. The 2019 materials by design roadmap. J. Phys. D Appl. Phys. 2018, 52, 013001. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.A.; Ahammad, N.A.; Awan, A.U.; Hussein, A.K.; Gamaoun, F.; Tag-ElDin, E.M.; Ali, B. Insight into the Role of Nanoparticles Shape Factors and Diameter on the Dynamics of Rotating Water-Based Fluid. Nanomaterials 2022, 12, 2801. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Babucci, M.; Casey, W.H.; Gates, B.C. The Surface Chemistry of Metal Oxide Clusters: From Metal–Organic Frameworks to Minerals. ACS Cent. Sci. 2020, 6, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, Y.; Roy, V.A.L.; Han, S. Evolutionary Metal Oxide Clusters for Novel Applications: Toward High-Density Data Storage in Nonvolatile Memories. Adv. Mater. 2017, 30, 1703950. [Google Scholar] [CrossRef]
- Li, X.-N.; Wang, L.-N.; Mou, L.-H.; He, S.-G. Catalytic CO Oxidation by Gas-Phase Metal Oxide Clusters. J. Phys. Chem. A 2019, 123, 9257–9267. [Google Scholar] [CrossRef] [PubMed]
- You, M.-H.; Li, M.-H.; Li, H.-H.; Chen, Y.; Lin, M.-J. The impact of metal cations on the photochemical properties of hybrid heterostructures with infinite alkaline-earth metal oxide clusters. Dalton Trans. 2019, 48, 17381–17387. [Google Scholar] [CrossRef]
- Shafi, M.A.; Bouich, A.; Fradi, K.; Guaita, J.M.; Khan, L.; Mari, B. Effect of deposition cycles on the properties of ZnO thin films deposited by spin coating method for CZTS-based solar cells. Optik 2022, 258, 168854. [Google Scholar] [CrossRef]
- Zemski, K.A.; Justes, D.R.; Castleman, A.W., Jr. ChemInform Abstract: Studies of Metal Oxide Clusters: Elucidating Reactive Sites Responsible for the Activity of Transition Metal Oxide Catalysts. ChemInform 2010, 33, 6136–6148. [Google Scholar] [CrossRef]
- Yu, B.Y.; Kwak, S.-Y. Carbon quantum dots embedded with mesoporous hematite nanospheres as efficient visible light-active photocatalysts. J. Mater. Chem. 2012, 22, 8345–8353. [Google Scholar] [CrossRef]
- Shinde, S.S.; Bansode, R.A.; Bhosale, C.H.; Rajpure, K.Y. Physical properties of hematite α-Fe2O3 thin films: Application to photoelectrochemical solar cells. J. Semicond. 2011, 32, 013001. [Google Scholar] [CrossRef]
- Pellin, M.J.; Riha, S.C.; Tyo, E.C.; Kwon, G.; Libera, J.A.; Elam, J.W.; Seifert, S.; Lee, S.; Vajda, S. Water Oxidation by Size-Selected Co27 Clusters Supported on Fe2O3. ChemSusChem 2016, 9, 3005–3011. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Ahmed, O.H.; Jiang, Z.T.; Dlugogorski, B.Z. Thermal recycling of brominated flame retardants with Fe2O3. J. Phys. Chem. A 2016, 120, 6039–6047. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.D.; Lee, S.W.; Kim, S.O.; Lee, J.K. Facile synthesis of carbon layer-entangled Fe2O3 clusters as anode materials for improved Li-ion batteries. J. Power Sources 2013, 244, 575–580. [Google Scholar] [CrossRef]
- Alaei, S.; Jalili, S.; Erkoc, S. Study of Electronic and Magnetic Properties of (Fe2O3) n Clusters Using Density Functional Theory. Quantum Matter 2016, 5, 607–611. [Google Scholar] [CrossRef]
- Shahpari, M.; Behjat, A.; Khajaminian, M.; Torabi, N. The influence of morphology of hematite (α-Fe2O3) counter electrodes on the efficiency of dye-sensitized solar cells. Sol. Energy 2015, 119, 45–53. [Google Scholar] [CrossRef]
- Sivula, K.; Le Formal, F.; Grätzel, M. Solar Water Splitting: Progress Using Hematite (α-Fe2O3) Photoelectrodes. ChemSusChem 2011, 4, 432–449. [Google Scholar] [CrossRef] [PubMed]
- Najaf, Z.; Nguyen, D.L.T.; Chae, S.Y.; Joo, O.S.; Shah, A.-u.-H.A.; Vo, D.-V.N.; Nguyen, V.-H.; Van Le, Q.; Rahman, G. Recent trends in development of hematite (α-Fe2O3) as an efficient photoanode for enhancement of photoelectrochemical hydrogen production by solar water splitting. Int. J. Hydrogen Energy 2021, 46, 23334–23357. [Google Scholar] [CrossRef]
- Ren, D.; Gui, K.; Gu, S.; Wei, Y. Mechanism of improving the SCR NO removal activity of Fe2O3 catalyst by doping Mn. J. Alloys Compd. 2021, 867, 158787. [Google Scholar] [CrossRef]
- Zhang, K.; Dong, T.; Xie, G.; Guan, L.; Guo, B.; Xiang, Q.; Dai, Y.; Tian, L.; Batool, A.; Jan, S.U.; et al. Sacrificial Interlayer for Promoting Charge Transport in Hematite Photoanode. ACS Appl. Mater. Interfaces 2017, 9, 42723–42733. [Google Scholar] [CrossRef]
- Fradi, K.; Bouich, A.; Slimi, B.; Chtourou, R. Towards improving the optoelectronics properties of MAPbI3(1−x)B3x/ZnO heterojunction by bromine doping. Optik 2021, 249, 168283. [Google Scholar] [CrossRef]
- Rakhshani, A.E. Preparation, characteristics and photovoltaic properties of cuprous oxide—A review. Solid-State Electron. 1986, 29, 7–17. [Google Scholar] [CrossRef]
- Grossiord, N.; Kroon, J.M.; Andriessen, R.; Blom, P.W. Degradation mechanisms in organic photovoltaic devices. Org. Electron. 2012, 13, 432–456. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Zhang, J.; Peng, T. New insight into the enhanced photocatalytic activity of N-, C- and S-doped ZnO photocatalysts. Appl. Catal. B Environ. 2016, 181, 220–227. [Google Scholar] [CrossRef]
- Pesci, F.M.; Wang, G.; Klug, D.R.; Li, Y.; Cowan, A.J. Efficient suppression of electron–hole recombination in ox-ygen-deficient hydrogen-treated TiO2 nanowires for photoelectrochemical water splitting. J. Phys. Chem. C 2013, 117, 25837–25844. [Google Scholar] [CrossRef] [PubMed]
- Abuilaiwi, F.A.; Awais, M.; Qazi, U.Y.; Afzal, A. Al3+ doping reduces the electron/hole recombination in photo-luminescent copper ferrite (CuFe2−xAlxO4) nanocrystallites. Boletín De La Soc. Española De Cerámica Y Vidr. 2022, 61, 252–262. [Google Scholar] [CrossRef]
- Sun, H.; Cao, Y.; Feng, L.; Chen, Y. Immobilizing photogenerated electrons from graphitic carbon nitride for an improved visible-light photocatalytic activity. Sci. Rep. 2016, 6, 22808. [Google Scholar] [CrossRef]
- Te Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Vicent-Luna, J.M.; Apergi, S.; Tao, S. Efficient Computation of Structural and Electronic Properties of Halide Per-ovskites Using Density Functional Tight Binding: GFN1−xTB Method. J. Chem. Inf. Modeling 2021, 61, 4415–4424. [Google Scholar] [CrossRef]
- Eom, T.; Kim, W.J.; Lim, H.K.; Han, M.H.; Han, K.H.; Lee, E.-K.; Lebègue, S.; Hwang, Y.J.; Min, B.K.; Kim, H. Cluster Expansion Method for Simu-lating Realistic Size of Nanoparticle Catalysts with an Application in CO2 Electroreduction. J. Phys. Chem. C 2018, 122, 9245–9254. [Google Scholar] [CrossRef]
- Korzhavyi, P.; Abrikosov, I.A.; Johansson, B.; Ruban, A.V.; Skriver, H.L. First-principles calculations of the vacancy formation energy in transition and noble metals. Phys. Rev. B 1999, 59, 11693–11703. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, J.; Smith, J.; Rose, J. Universal Binding Energy Relations in Metallic Adhesion. Tribol. Ser. 1981, 7, 19–30. [Google Scholar] [CrossRef]
- Majid, A.; Zahid, S.; Khan, S.U.D. Theoretical study of (TM) FeO3 (TM = 3d transition metals) molecular clusters. J. Nanoparticle Res. 2020, 22, 1–20. [Google Scholar] [CrossRef]
- Li, K.; Xue, D. Estimation of Electronegativity Values of Elements in Different Valence States. J. Phys. Chem. A 2006, 110, 11332–11337. [Google Scholar] [CrossRef] [PubMed]
- Tiest, W.M.B.; Kappers, A.M. Physical Aspects of Softness Perception. In Multisensory Softness; Springer: Berlin, Germany, 2014; pp. 3–15. [Google Scholar] [CrossRef]
- Vogiatzis, K.D.; Polynski, M.V.; Kirkland, J.K.; Townsend, J.; Hashemi, A.; Liu, C.; Pidko, E.A. Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities. Chem. Rev. 2018, 119, 2453–2523. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Bhattacharyya, N.; Bandyopadhyay, D. Architecture, electronic structure and stability of TM@Ge(n)(TM = Ti, Zr and Hf; n = 1–20) clusters: A density functional modeling. J. Mol. Modeling 2012, 18, 405–418. [Google Scholar] [CrossRef]
- Khan, I.; Jalilov, A.; Fujii, K.; Qurashi, A. Quasi-1D Aligned Nanostructures for Solar-Driven Water Splitting Ap-plications: Challenges, Promises, and Perspectives. Solar RRL 2021, 5, 2000741. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef]
- Merazga, A.; Al-Zahrani, J.; Al-Baradi, A.; Omer, B.; Badawi, A.; Al-Omairy, S. Optical band-gap of reduced gra-phene oxide/TiO2 composite and performance of associated dye-sensitized solar cells. Mater. Sci. Eng. B 2020, 259, 114581. [Google Scholar] [CrossRef]
- Hamadanian, M.; Jabbari, V.; Gravand, A.; Asad, M. Band gap engineering of TiO2 nanostructure-based dye solar cells (DSCs) fabricated via electrophoresis. Surf. Coatings Technol. 2012, 206, 4531–4538. [Google Scholar] [CrossRef]
- Arifin, Z.; Suyitno, S.; Hadi, S.; Sutanto, B. Improved performance of dye-sensitized solar cells with TiO2 nanopar-ticles/Zn-doped TiO2 hollow fiber photoanodes. Energies 2018, 11, 2922. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
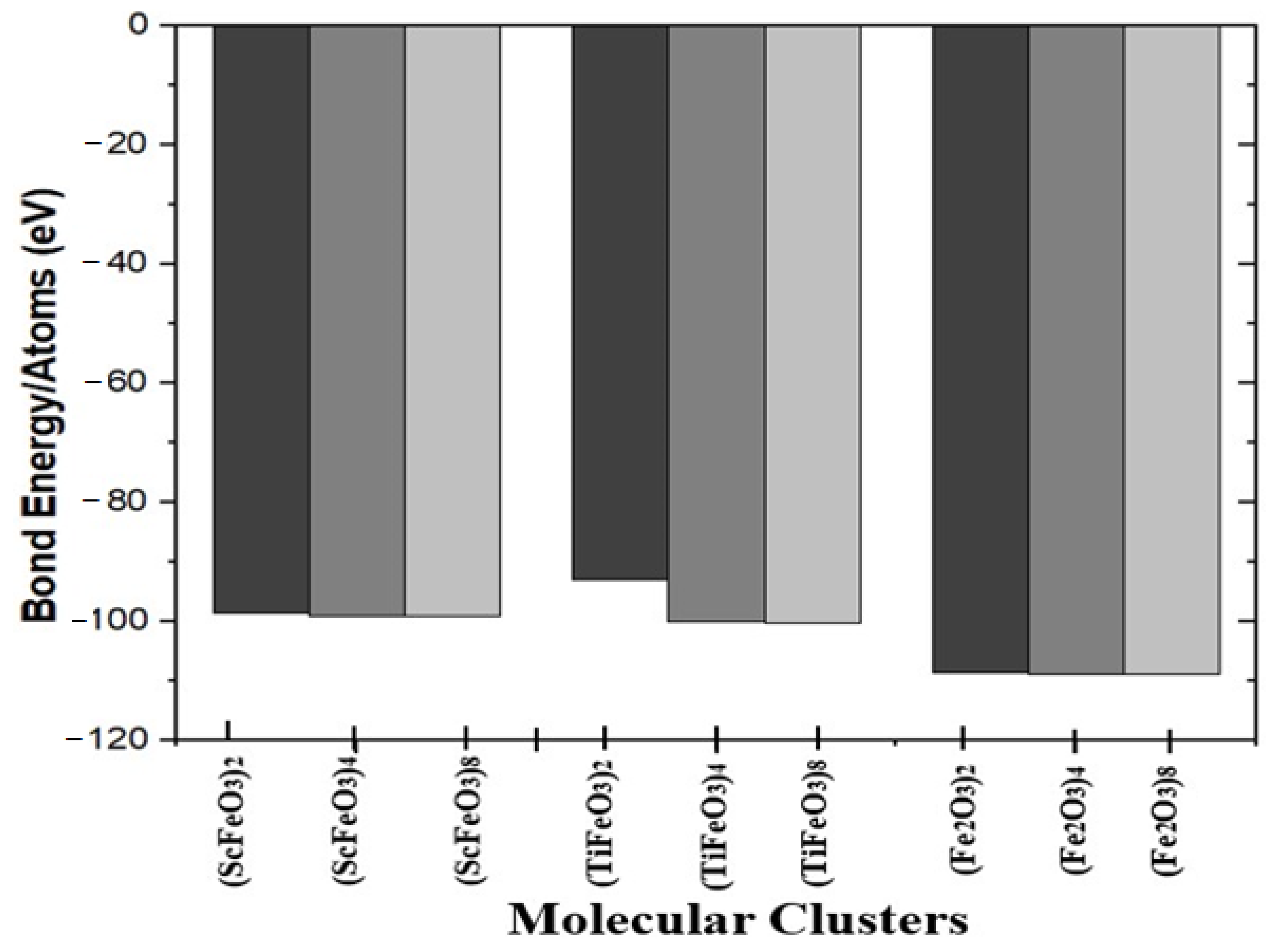
| Molecular Clusters | Bond Energy (eV) | Binding Energy (eV) | Formation Energy EF(eV) |
|---|---|---|---|
| (ScFeO3)2 | −987.20 | 25.114 | −25.114 |
| (ScFeO3)4 | −1980.7 | 56.538 | −56.538 |
| (ScFeO3)8 | −3968.8 | 120.49 | −120.49 |
| (TiFeO3)2 | −930.047 | 24.394 | −24.394 |
| (TiFeO3)4 | −2002.7 | 65.438 | −65.438 |
| (TiFeO3)8 | −4014.7 | 140.14 | −140.14 |
| (Fe2O3)2 | −1087.5 | 33.638 | −33.638 |
| (Fe2O3)4 | −2177.1 | 62.256 | −62.256 |
| (Fe2O3)8 | −4357.1 | 141.47 | −141.47 |
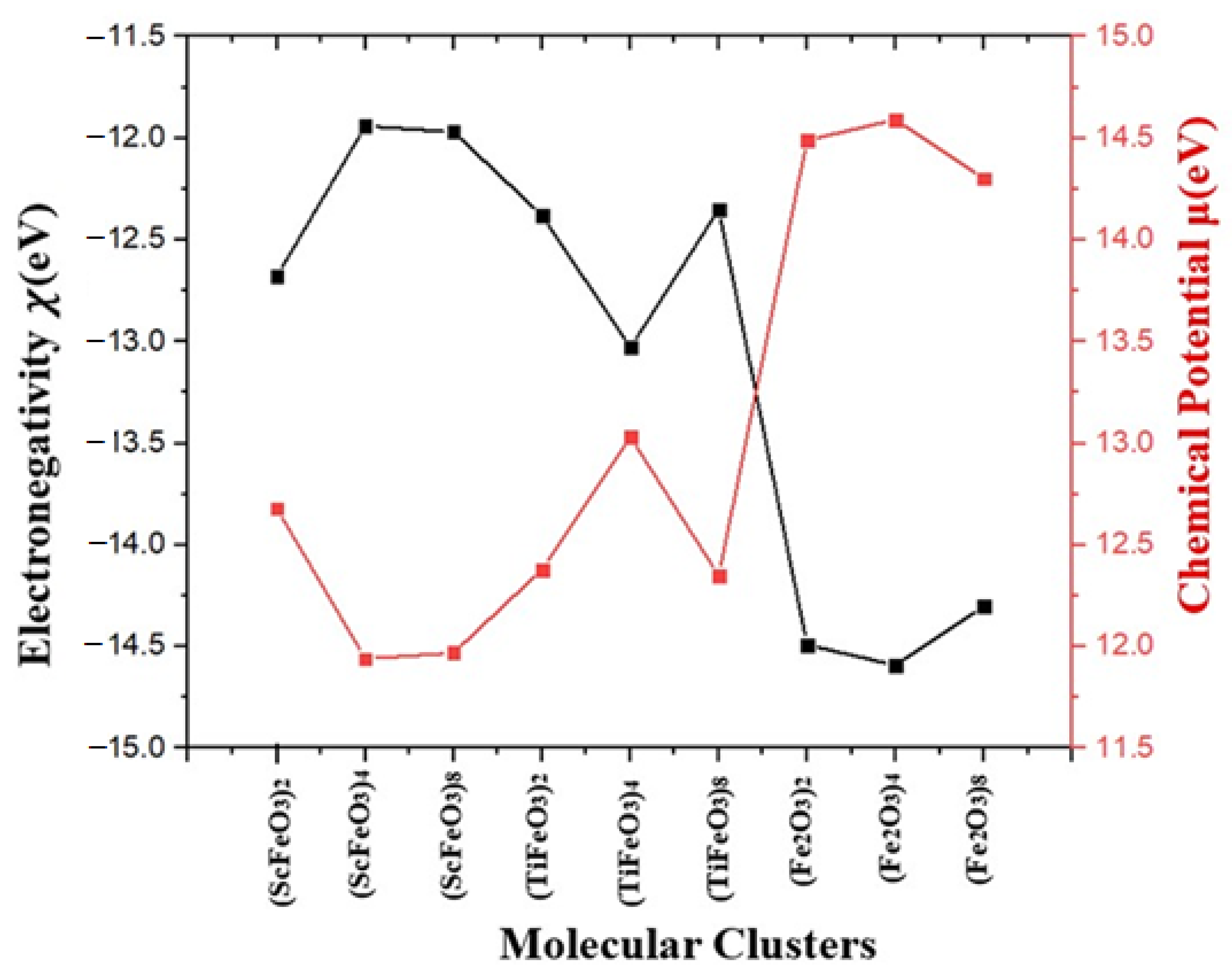
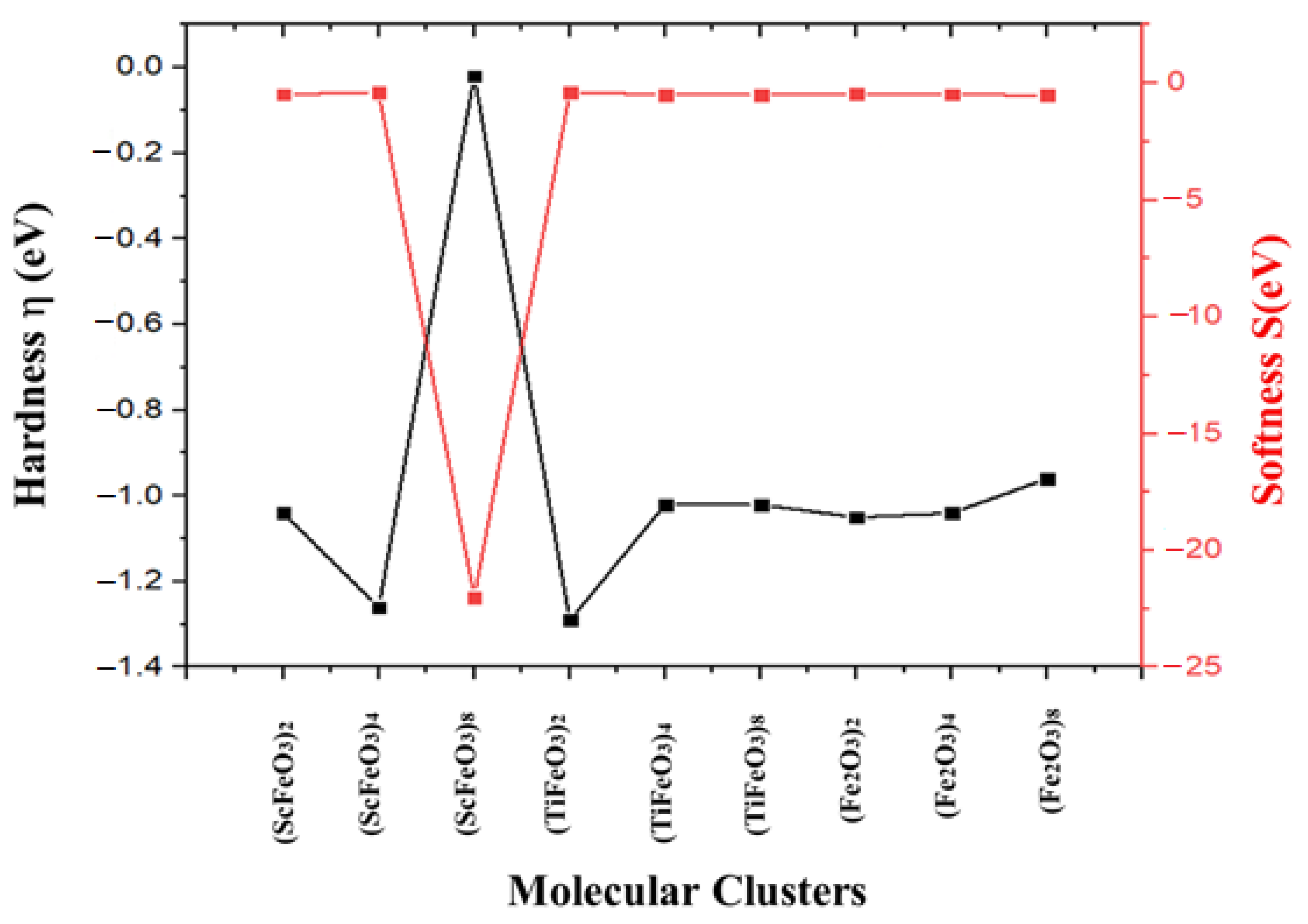
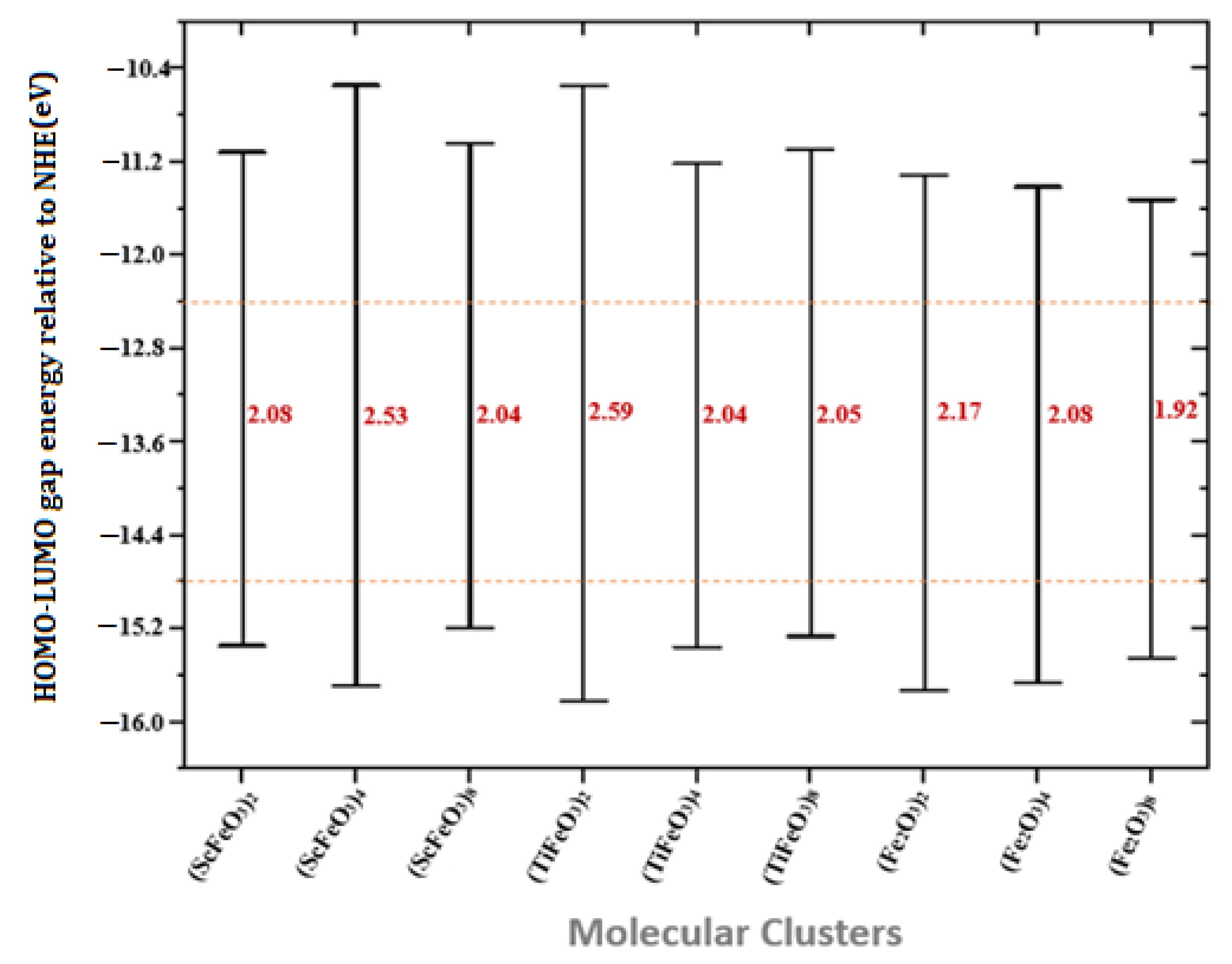
| Molecular Clusters | EA (eV) | IP (eV) | B.G. (eV) | η (eV) | S (eV) | (eV) | Χ (eV) |
|---|---|---|---|---|---|---|---|
| (ScFeO3)2 | −11.64 | −13.72 | 2.08 | −1.04 | −0.48 | 12.68 | −12.68 |
| (ScFeO3)4 | −10.68 | −13.21 | 2.53 | −1.26 | −0.39 | 11.94 | −11.94 |
| (ScFeO3)8 | −10.95 | −12.99 | 2.04 | −0.02 | −22.0 | 11.97 | −11.97 |
| (TiFeO3)2 | −11.09 | −13.68 | 2.59 | −1.29 | −0.38 | 12.38 | −12.38 |
| (TiFeO3)4 | −12.01 | −14.05 | 2.04 | −1.02 | −0.49 | 13.03 | −13.03 |
| (TiFeO3)8 | −11.33 | −13.38 | 2.05 | −1.02 | −0.49 | 12.35 | −12.35 |
| (Fe2O3)2 | −13.41 | −15.58 | 2.17 | −1.05 | −0.47 | 14.49 | −14.49 |
| (Fe2O3)4 | −13.55 | −15.64 | 2.08 | −1.04 | −0.48 | 14.59 | −14.59 |
| (Fe2O3)8 | −13.34 | −15.27 | 1.92 | −0.96 | −0.52 | 14.30 | −14.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majid, A.; Arif, S.; Younes, T.M.; Alkhedher, M.; ElDin, S.M. DFT Study of Heteronuclear (TMFeO3)x Molecular Clusters (Where TM = Sc, Ti, Fe and x = 2, 4, 8) for Photocatalytic and Photovoltaic Applications. Energies 2022, 15, 7253. https://doi.org/10.3390/en15197253
Majid A, Arif S, Younes TM, Alkhedher M, ElDin SM. DFT Study of Heteronuclear (TMFeO3)x Molecular Clusters (Where TM = Sc, Ti, Fe and x = 2, 4, 8) for Photocatalytic and Photovoltaic Applications. Energies. 2022; 15(19):7253. https://doi.org/10.3390/en15197253
Chicago/Turabian StyleMajid, Abdul, Sidra Arif, Tariq M. Younes, Mohammad Alkhedher, and Sayed M. ElDin. 2022. "DFT Study of Heteronuclear (TMFeO3)x Molecular Clusters (Where TM = Sc, Ti, Fe and x = 2, 4, 8) for Photocatalytic and Photovoltaic Applications" Energies 15, no. 19: 7253. https://doi.org/10.3390/en15197253
APA StyleMajid, A., Arif, S., Younes, T. M., Alkhedher, M., & ElDin, S. M. (2022). DFT Study of Heteronuclear (TMFeO3)x Molecular Clusters (Where TM = Sc, Ti, Fe and x = 2, 4, 8) for Photocatalytic and Photovoltaic Applications. Energies, 15(19), 7253. https://doi.org/10.3390/en15197253