
Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes



Abstract
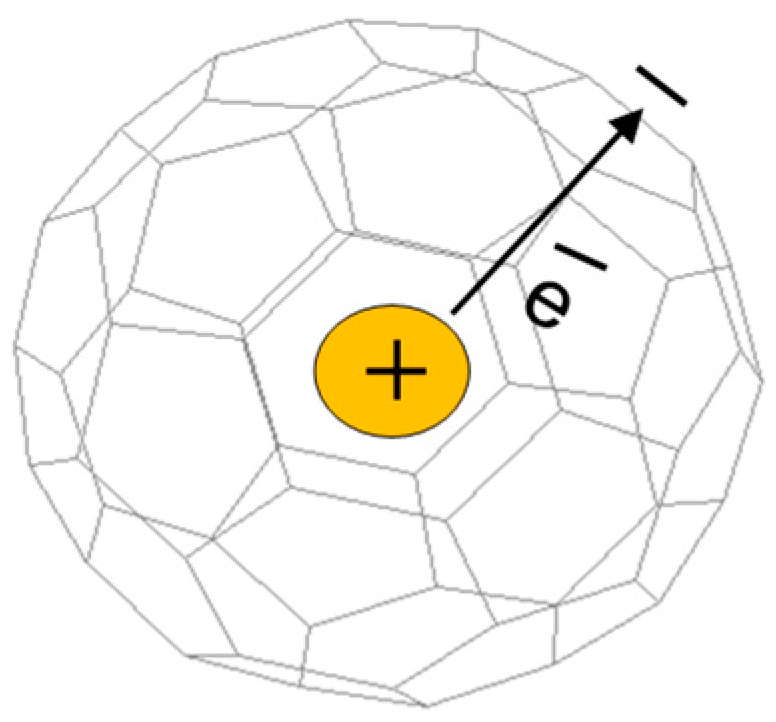
:1. Introduction
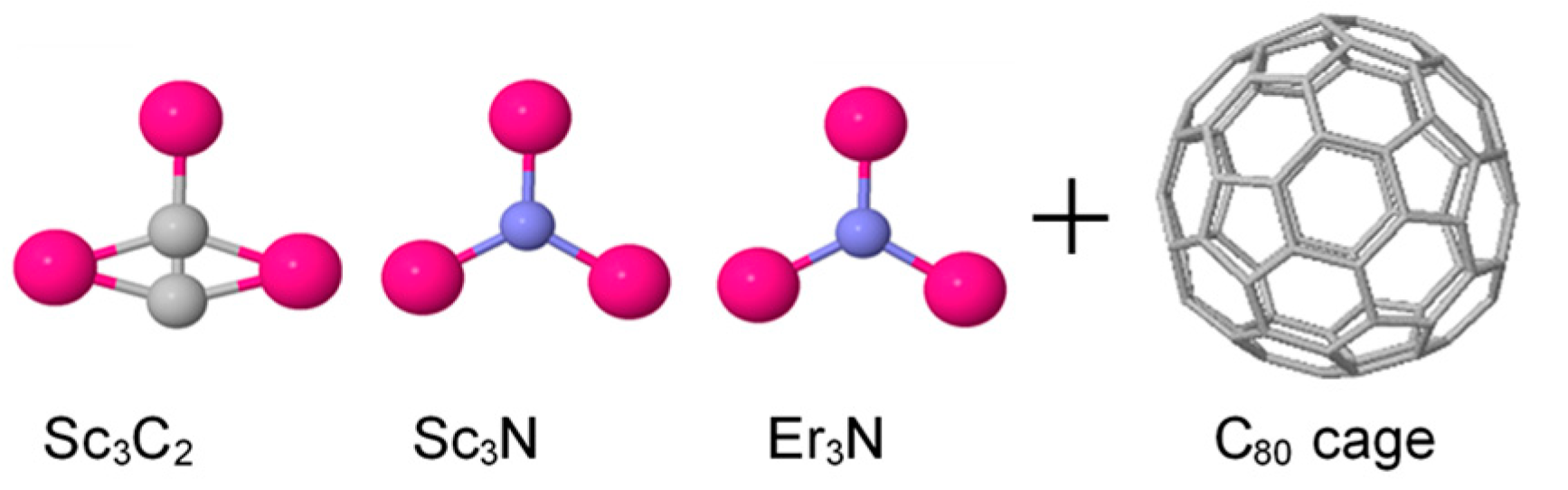
2. Computational Methods
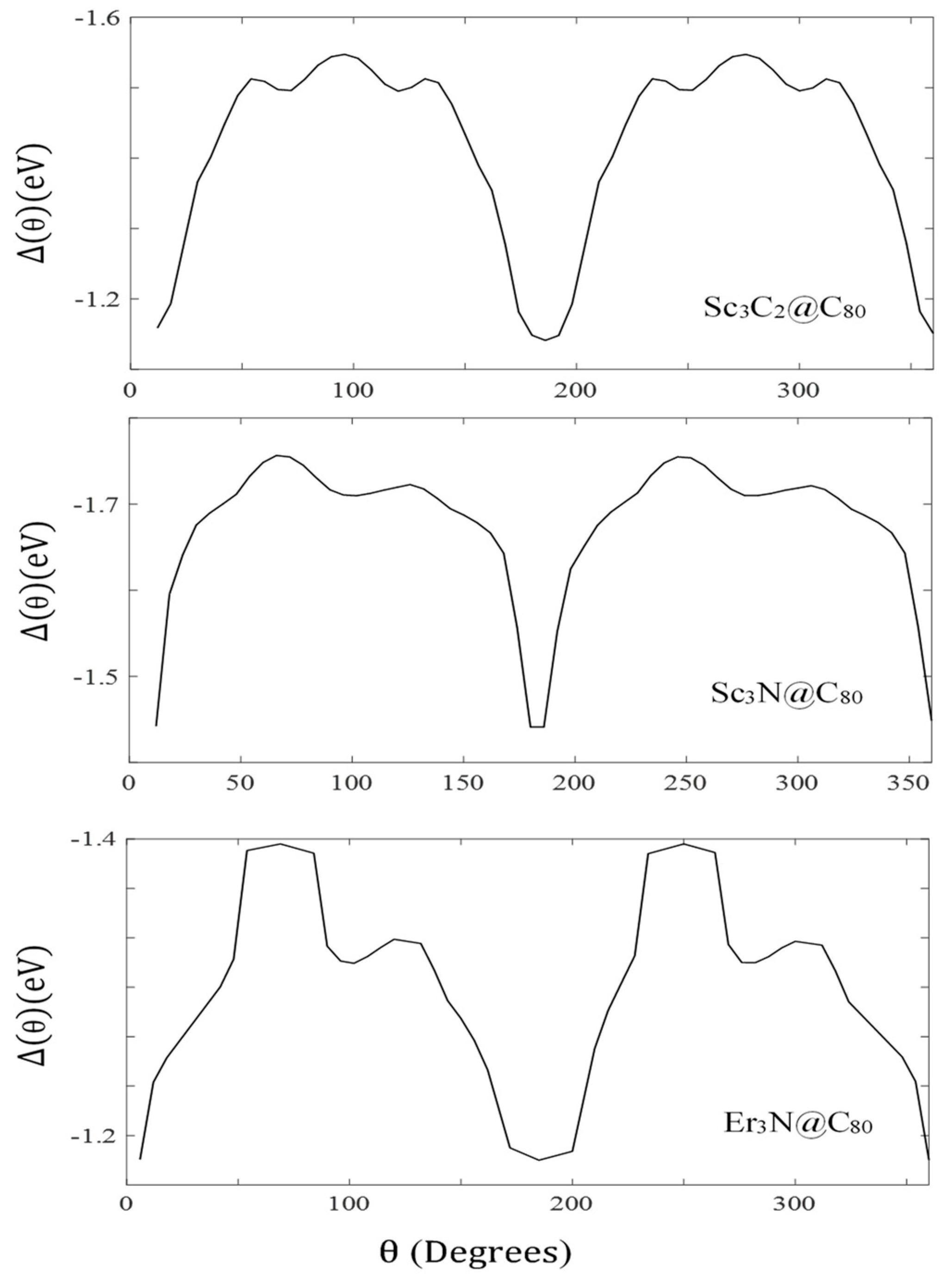
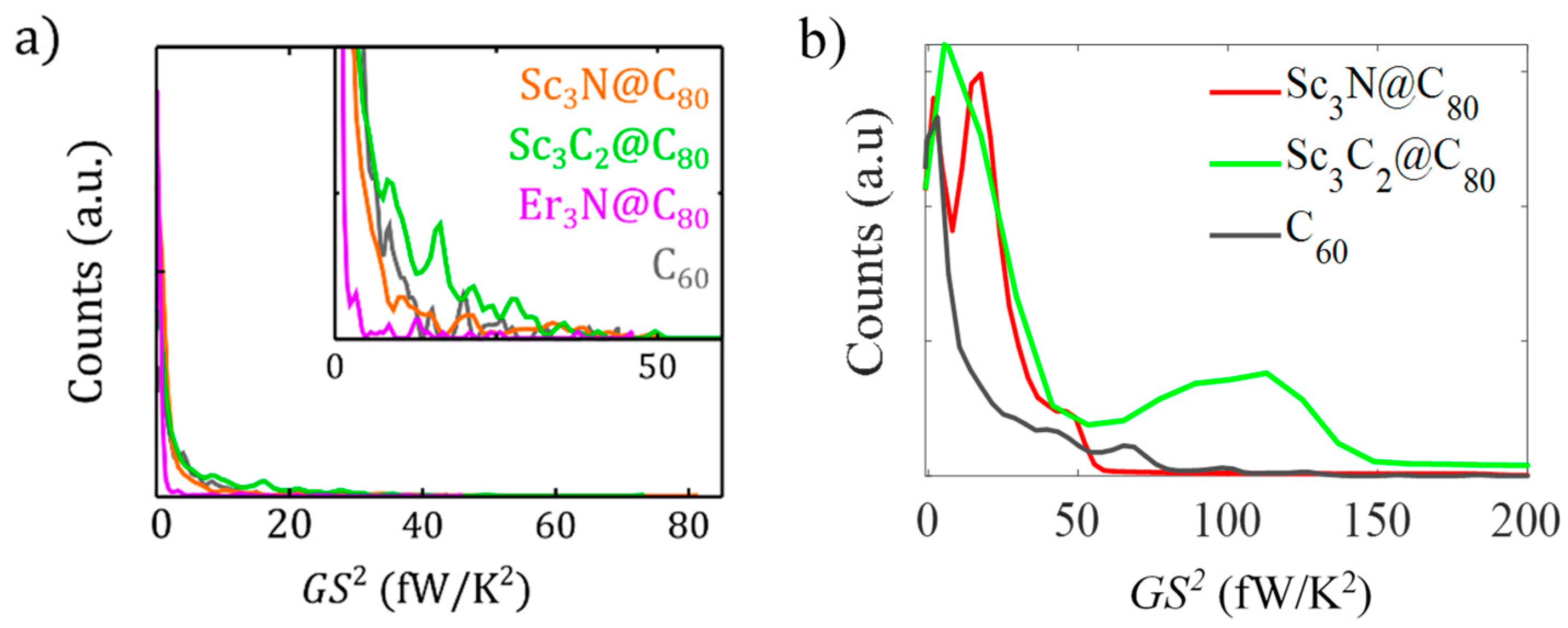
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Soos, Z.G. Theory of π-molecular charge-transfer crystals. Annu. Rev. Phys. Chem. 1974, 25, 121–153. [Google Scholar] [CrossRef]
- Bauer, C.; Teuscher, J.; Brauer, J.C.; Punzi, A.; Marchioro, A.; Ghadiri, E.; De Jonghe, J.; Wielopolski, M.; Banerji, N.; Moser, J.-E. Dynamics and mechanisms of interfacial photoinduced electron transfer processes of third generation photovoltaics and photocatalysis. CHIMIA Int. J. Chem. 2011, 65, 704–709. [Google Scholar] [CrossRef]
- Günes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated polymer-based organic solar cells. Chem. Rev. 2007, 107, 1324–1338. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Méndez-Hernández, D.D.; Tejeda-Ferrari, M.E.; Teillout, A.-L.; Llansola-Portolés, M.J.; Kodis, G.; Poluektov, O.G.; Rajh, T.; Mujica, V.; Groy, T.L. A bioinspired redox relay that mimics radical interactions of the Tyr–His pairs of photosystem. Nat. Chem. 2014, 6, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Aviram, A.; Ratner, M.A. Molecular rectifiers. Bull. Am. Phys. Soc. 1974, 19, 341. [Google Scholar] [CrossRef]
- Herrer, L.; Ismael, A.; Martin, S.; Milan, D.C.; Serrano, J.L.; Nichols, R.J.; Lambert, C.; Cea, P. Single molecule vs. large area design of molecular electronic devices incorporating an efficient 2-aminepyridine double anchoring group. Nanoscale 2019, 11, 15871–15880. [Google Scholar] [CrossRef]
- Al-Khaykanee, M.K.; Ismael, A.K.; Grace, I.; Lambert, C.J. Oscillating Seebeck coefficients in π-stacked molecular junctions. Rsc Adv. 2018, 8, 24711–24715. [Google Scholar] [CrossRef]
- Bockrath, M.; Cobden, D.H.; McEuen, P.L.; Chopra, N.G.; Zettl, A.; Thess, A.; Smalley, R.E. Single-electron transport in ropes of carbon nanotubes. Science 1997, 275, 1922–1925. [Google Scholar] [CrossRef]
- Ismael, A.K.; Lambert, C.J. Single-molecule conductance oscillations in alkane rings. J. Mater. Chem. C 2019, 7, 6578–6581. [Google Scholar] [CrossRef]
- Romaner, L.; Heimel, G.; Brédas, J.-L.; Gerlach, A.; Schreiber, F.; Johnson, R.L.; Zegenhagen, J.; Duhm, S.; Koch, N.; Zojer, E. Impact of bidirectional charge transfer and molecular distortions on the electronic structure of a metal-organic interface. Phys. Rev. Lett. 2007, 99, 256801. [Google Scholar] [CrossRef]
- Bennett, T.L.; Alshammari, M.; Au-Yong, S.; Almutlg, A.; Wang, X.; Wilkinson, L.A.; Albrecht, T.; Jarvis, S.P.; Cohen, L.F.; Ismael, A. Multi-component self-assembled molecular-electronic films: Towards new high-performance thermoelectric systems. Chem. Sci. 2022, 13, 5176–5185. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Chen, G.; Perry, J.W.; Goddard, W.A., III. Valence-bond charge-transfer model for nonlinear optical properties of charge-transfer organic molecules. J. Am. Chem. Soc. 1994, 116, 10679–10685. [Google Scholar] [CrossRef]
- Gorczak, N.; Renaud, N.; Tarkuç, S.; Houtepen, A.J.; Eelkema, R.; Siebbeles, L.D.; Grozema, F.C. Charge transfer versus molecular conductance: Molecular orbital symmetry turns quantum interference rules upside down. Chem. Sci. 2015, 6, 4196–4206. [Google Scholar] [CrossRef] [PubMed]
- Closs, G.L.; Miller, J.R. Intramolecular long-distance electron transfer in organic molecules. Science 1988, 240, 440–447. [Google Scholar] [CrossRef]
- Sukegawa, J.; Schubert, C.; Zhu, X.; Tsuji, H.; Guldi, D.M.; Nakamura, E. Electron transfer through rigid organic molecular wires enhanced by electronic and electron–vibration coupling. Nat. Chem. 2014, 6, 899–905. [Google Scholar] [CrossRef]
- Deibel, C.; Strobel, T.; Dyakonov, V. Role of the charge transfer state in organic donor–acceptor solar cells. Adv. Mater. 2010, 22, 4097–4111. [Google Scholar] [CrossRef]
- Otero, R.; de Parga, A.V.; Gallego, J.M. Electronic, structural and chemical effects of charge-transfer at organic/inorganic interfaces. Surf. Sci. Rep. 2017, 72, 105–145. [Google Scholar] [CrossRef]
- Kollmannsberger, M.; Rurack, K.; Resch-Genger, U.; Rettig, W.; Daub, J. Design of an efficient charge-transfer processing molecular system containing a weak electron donor: Spectroscopic and redox properties and cation-induced fluorescence enhancement. Chem. Phys. Lett. 2000, 329, 363–369. [Google Scholar] [CrossRef]
- Wörner, H.J.; Arrell, C.A.; Banerji, N.; Cannizzo, A.; Chergui, M.; Das, A.K.; Hamm, P.; Keller, U.; Kraus, P.M.; Liberatore, E. Charge migration and charge transfer in molecular systems. Struct. Dyn. 2017, 4, 061508. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Guerra, C.F.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D.J.J.o.P.C.M. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Davidson, R.J.; Milan, D.C.; Al-Owaedi, O.A.; Ismael, A.K.; Nichols, R.J.; Higgins, S.J.; Lambert, C.J.; Yufit, D.S.; Beeby, A. Conductance of ‘bare-bones’ tripodal molecular wires. RSC Adv. 2018, 8, 23585–23590. [Google Scholar] [CrossRef] [PubMed]
- Markin, A.; Ismael, A.K.; Davidson, R.J.; Milan, D.C.; Nichols, R.J.; Higgins, S.J.; Lambert, C.J.; Hsu, Y.-T.; Yufit, D.S.; Beeby, A. Conductance Behavior of Tetraphenyl-Aza-BODIPYs. J. Phys. Chem. C 2020, 124, 6479–6485. [Google Scholar] [CrossRef]
- Kobko, N.; Dannenberg, J. Dannenberg. Effect of basis set superposition error (BSSE) upon ab initio calculations of organic transition states. J. Phys. Chem. A 2001, 105, 1944–1950. [Google Scholar] [CrossRef]
- Sherrill, C.D. Counterpoise Correction and Basis Set Superposition Error; School of Chemistry and Biochemistry, Georgia Institute of Technology: Atlanta, Georgia, 2010. [Google Scholar]
- Sinnokrot, M.O.; Valeev, E.F.; Sherrill, C.D. Estimates of the ab initio limit for π− π interactions: The benzene dimer. J. Am. Chem. Soc. 2002, 124, 10887–10893. [Google Scholar] [CrossRef]
- Ismael, A.K.; Rincón-García, L.; Evangeli, C.; Dallas, P.; Alotaibi, T.; Al-Jobory, A.A.; Rubio-Bollinger, G.; Porfyrakis, K.; Agraït, N.; Lambert, C.J. Exploring seebeck-coefficient fluctuations in endohedral-fullerene, single-molecule junctions. Nanoscale Horiz. 2022, 7, 616–625. [Google Scholar] [CrossRef]
- Akkermans, E.; Montambaux, G. Mesoscopic Physics of Electrons and Photons; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Ismael, A.K.; Al-Jobory, A.; Grace, I.; Lambert, C.J. Discriminating single-molecule sensing by crown-ether-based molecular junctions. J. Chem. Phys. 2017, 146, 064704. [Google Scholar] [CrossRef]
- Ismael, A.K.; Grace, I.; Lambert, C.J. Increasing the thermopower of crown-ether-bridged anthraquinones. Nanoscale 2015, 7, 17338–17342. [Google Scholar] [CrossRef] [PubMed]
- Ismael, A.K.; Grace, I.; Lambert, C.J. Connectivity dependence of Fano resonances in single molecules. Phys. Chem. Chem. Phys. 2017, 19, 6416–6421. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ismael, A.; Ning, S.; Althobaiti, H.; Al-Jobory, A.; Girovsky, J.; Astier, H.P.; O'Driscoll, L.J.; Bryce, M.R.; Lambert, C.J. Electrostatic Fermi level tuning in large-scale self-assembled monolayers of oligo (phenylene–ethynylene) derivatives. Nanoscale Horiz. 2022, 7, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L.A.; Bennett, T.L.; Grace, I.M.; Hamill, J.; Wang, X.; Au-Yong, S.; Ismael, A.; Jarvis, S.P.; Hou, S.; Albrecht, T. Assembly, structure and thermoelectric properties of 1,1′-dialkynylferrocene ‘hinges’. Chem. Sci. 2022, 13, 8380–8387. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Al-Jobory, A.; Zhang, Q.-C.; Cao, W.; Alshehab, A.; Qu, K.; Alotaibi, T.; Chen, H.; Liu, J.; Ismael, A.K. Highly insulating alkane rings with destructive σ-interference. Sci. China Chem. 2022, 65, 1822–1828. [Google Scholar] [CrossRef]
- Rincón-García, L.; Ismael, A.K.; Evangeli, C.; Grace, I.; Rubio-Bollinger, G.; Porfyrakis, K.; Agraït, N.; Lambert, C.J. Molecular design and control of fullerene-based bi-thermoelectric materials. Nat. Mater. 2016, 15, 289–293. [Google Scholar] [CrossRef]
- Lu, J.; Nagase, S.; Zhang, X.; Wang, D.; Ni, M.; Maeda, Y.; Wakahara, T.; Nakahodo, T.; Tsuchiya, T.; Akasaka, T. Selective interaction of large or charge-transfer aromatic molecules with metallic single-wall carbon nanotubes: Critical role of the molecular size and orientation. J. Am. Chem. Soc. 2006, 128, 5114–5118. [Google Scholar] [CrossRef]
- Ismael, A.; Al-Jobory, A.; Wang, X.; Alshehab, A.; Almutlg, A.; Alshammari, M.; Grace, I.; Benett, T.L.; Wilkinson, L.A.; Robinson, B.J. Molecular-scale thermoelectricity: As simple as ‘ABC’. Nanoscale Adv. 2020, 2, 5329–5334. [Google Scholar] [CrossRef]
- Lee, S.K.; Buerkle, M.; Yamada, R.; Asai, Y.; Tada, H. Thermoelectricity at the molecular scale: A large Seebeck effect in endohedral metallofullerenes. Nanoscale 2015, 7, 20497–20502. [Google Scholar] [CrossRef]
- Balachandran, J.; Reddy, P.; Dunietz, B.D.; Gavini, V. End-group-induced charge transfer in molecular junctions: Effect on electronic-structure and thermopower. J. Phys. Chem. Lett. 2012, 3, 1962–1967. [Google Scholar] [CrossRef]
- Adams, D.M.; Brus, L.; Chidsey, C.E.; Creager, S.; Creutz, C.; Kagan, C.R.; Kamat, P.V.; Lieberman, M.; Lindsay, S.; Marcus, R.A. Charge transfer on the nanoscale: Current status. J. Phys. Chem. B 2003, 107, 6668–6697. [Google Scholar] [CrossRef]
- Liu, S.-X.; Ismael, A.K.; Al-Jobory, A.; Lambert, C.J. Signatures of Room-Temperature Quantum Interference in Molecular Junctions. Acc. Chem. Res. 2023, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Alshehab, A.; Ismael, A.K. Impact of the terminal end-group on the electrical conductance in alkane linear chains. RSC Adv. 2023, 13, 5869–5873. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metallic Moiety | Mulliken | Hirshfeld | Voronoi | |||
|---|---|---|---|---|---|---|
| moiety | cage | moiety | cage | moiety | cage | |
| Sc3C2 | +1.40 | −1.14 | +1.15 | −0.83 | +1.06 | −0.72 |
| C2 | (−0.26) | - | (−0.32) | - | (−0.34) | - |
| Sc3N | +1.50 | −1.26 | +1.31 | −0.98 | +1.27 | −0.96 |
| N | (−0.24) | - | (−0.33) | - | (−0.31) | - |
| Er3N | +6.96 | −5.14 | +7.48 | −6.14 | +7.14 | −5.82 |
| N | (−1.82) | - | (−1.34) | - | (−1.32) | - |
| Moiety + Au | Mulliken | Hirshfeld | Voronoi | |||
|---|---|---|---|---|---|---|
| moiety | cage | moiety | cage | moiety | cage | |
| Sc3C2 | +1.33 | −1.39 | +0.93 | –0.81 | +0.97 | −0.79 |
| Au, C2 | (+0.3, −0.24) | - | (+0.2, −0.32) | - | (+0.18, −0.36) | - |
| Sc3N | +2.15 | −2.09 | +1.07 | –0.98 | +1.04 | –1.02 |
| Au, N | (+0.22, −0.28) | - | (+0.23, −0.32) | - | (+0.24, −0.26) | - |
| Er3N | +6.53 | −5.20 | +6.96 | –5.80 | +6.66 | −5.60 |
| Au, N | (+0.24, −1.57) | - | (+0.28, −1.44) | - | (+0.29, −1.35) | - |
| EMF Complex | |||
|---|---|---|---|
| Sc3C2@C80 | 0.0154 | 0.0113 | 0.0133 |
| Sc3N@C80 | 0.0163 | 0.0109 | 0.0119 |
| Er3N@C80 | 0.00378 | 0.00259 | 0.00268 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshammari, M.; Alotaibi, T.; Alotaibi, M.; Ismael, A.K. Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes. Energies 2023, 16, 4342. https://doi.org/10.3390/en16114342
Alshammari M, Alotaibi T, Alotaibi M, Ismael AK. Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes. Energies. 2023; 16(11):4342. https://doi.org/10.3390/en16114342
Chicago/Turabian StyleAlshammari, Majed, Turki Alotaibi, Moteb Alotaibi, and Ali K. Ismael. 2023. "Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes" Energies 16, no. 11: 4342. https://doi.org/10.3390/en16114342
APA StyleAlshammari, M., Alotaibi, T., Alotaibi, M., & Ismael, A. K. (2023). Influence of Charge Transfer on Thermoelectric Properties of Endohedral Metallofullerene (EMF) Complexes. Energies, 16(11), 4342. https://doi.org/10.3390/en16114342