1. Introduction
As rapid development and population increase have caused global environmental problems and depletion of fossil fuels, extensive efforts are devoted to renewable energy technologies to develop alternative energy resources and counteract global warming. Biomass has several strengths as an energy resource. Firstly, the net carbon emission from biomass is, at least theoretically, zero,
i.e., it is carbon-neutral, because the carbon included in biomass originated from atmospheric CO
2 as a result of photosynthesis of plants. Secondly, its sulfur content is very low compared to fossil fuels and hence it causes less air pollution upon combustion. Thirdly, it is renewable resource in that it is produced using solar energy. Due to these strengths, biomass is regarded as one of the most important future energy resources and the technologies for energy production from biomass have received large attention from all over the world [
1,
2].
Being a potential energy-producing biomass, reed is a C3 perennial grass with rich contents of lignin and cellulose. Wild reed that grows at Suncheon Bay Ecological Park (SBEP) is cut regularly to maintain the ecological equilibrium. Although some cut reed is recycled as construction materials, most of it is incinerated or landfilled. Therefore, finding an energy production method using SBEP wild reed is important in terms of both waste reduction and developing a new regional renewable energy resource.
Pyrolysis is a process in which organic matter is thermally converted to solid, liquid and gas products under a high-temperature oxygen-free atmosphere [
3]. In particular, fast pyrolysis was devised to maximize the liquid pyrolysis product, called bio-oil. In order to obtain high bio-oil yield from pyrolysis, short gas residence time in reactor and high temperature rise rate are required [
1].
Catalytic pyrolysis of biomass over acidic catalysts has attracted significant attention as a method of promoting the production of aliphatic and aromatic hydrocarbons [
4,
5]. Zeolite is an aluminosilicate material containing micropores. Having acid sites in its pores, zeolite is widely used in catalytic pyrolysis of biomass [
6,
7]. In a couple of previous studies, we reported the results of non-catalytic and catalytic fast pyrolysis of SBEP wild reed [
8,
9]. HZSM-5 zeolite catalysts were used for catalytic pyrolysis. Compared to non-catalytic pyrolysis, catalytic pyrolysis reduced the quantity of oxygenates in bio-oil considerably, while increasing the contents of furans and aromatics. This was attributed to the conversion of oxygenates to furans and aromatics on the Brønsted acid sites. A shortcoming of catalytic pyrolysis is the high price of the catalysts. Application of waste catalyst can reduce the cost of catalytic pyrolysis.
Fluidized catalytic cracking (FCC) catalyst, which is used in petrochemical refinery processes to reduce the molecular mass of oil species, is composed mostly of synthetic silica-alumina, containing 5–20 wt % of zeolite. In order to increase the hydrothermal stability of the catalyst, ultrastable Y (USY) zeolite, in which alkali metal ions of zeolite are ion-exchanged with H
+ or rare-earth metal cations, is usually used as the framework of FCC catalysts [
10]. After use, the waste FCC catalyst consisting mostly of Si and Al is usually used as raw material for cement production with low value-added.
In a previous study [
11], waste FCC catalyst was applied for the pyrolysis of polypropylene. The main species of the bio-oil obtained were found to be hydrocarbons in diesel range. Jang,
et al. [
12] activated waste FCC catalyst using HCl and NaOH solutions and used it for liquid-phase decomposition reaction of waste agricultural film. They reported that waste FCC catalyst can be an economical resource for recycling waste agricultural film although its catalytic activity was lower than commercial USY zeolite catalyst. These previous studies demonstrated the potential of waste FCC catalyst as a catalyst for biomass pyrolysis. To the best of our knowledge, however, the application of waste FCC catalyst for the pyrolysis of natural biomass has never been reported.
In this study, waste FCC catalyst was applied for the catalytic pyrolysis of wild reed. A zeolite catalyst HY was also used for comparison.
2. Experimental
2.1. Materials
Zeolite-based waste FCC catalyst and commercial HY zeolite catalyst were used in this study. HY with a SiO2/Al2O3 ratio of 5.1 was purchased from Zeolyst International. Waste FCC catalyst disposed from a petrochemical refinery process was soaked in acetone with a mass ratio of 1:10 and the mixture was stirred for 24 h, aimed at washing out residual organic materials (physical activation). After the acetone washing, it was dried for an hour in a 60 °C dryer. As another method of activation of waste FCC catalyst, the dried catalyst was calcined for 3 h in air using a 550 °C quartz tube reactor (chemical activation). All catalysts were pelletized into uniform size (1.5–2.5 mm) prior to use.
Thermo-gravimetric analysis (TGA) was performed using a pyris1 thermo-gravimetric analyzer (Perkin Elmer, Waltham, MA, USA) to evaluate the effects of physical and chemical activation treatments. During the analysis, temperature was increased at a rate of 30 °C/min from 30 °C to 700 °C. The specific surface area of catalysts was measured using Brunauer-Emmett-Teller (BET) method. After pre-treating catalyst samples in vacuum at 200 °C, the adsorption and desorption of nitrogen by the samples were measured at 77 K using a BELSORP-MINI system (BEL Japan Inc., Osaka, Japan) to create the sorption isotherms. The specific surface area was determined from the isotherms based on the BET method. The crystalline structure of the catalysts was examined using a high-power X-ray diffractometer (XRD).
Wild reed of SBEP was collected for the experiments of this study. Dried wild reed sample was pulverized and sieved into uniform diameter of 2 mm on average (1–3 mm). The sample particles were then dried in an oven at 105 ± 5 °C for 24 h.
2.2. Experiment and Analysis
The batch-type reactor used in the previous studies [
8,
9] was used for catalytic pyrolysis in this study. The reactor was a U-tube quartz reactor, with an internal volume of 50 mL. The furnace temperature was controlled using a proportional-integral-derivative controller with an uncertainty of ±5 °C. The reactor temperature was increased to the pyrolysis temperature instantly by inserting the reactor into pre-heated furnace.
The reactor was filled with a mixture of 1 g of reed sample and 1 or 10 g of catalyst. Pyrolysis was allowed to proceed at 500 °C with a nitrogen gas flowrate of 50 mL/min controlled at room temperature. Taking the volume expansion of carrier gas at 500 °C and the reactor volume reduction due to biomass and catalyst into account, the residence time in the reactor was 20 s and 14 s with the catalyst mass of 1 g and 10 g, respectively. The reactor was purged with nitrogen gas for 5 min before the pyrolysis experiment to make the reactor atmosphere oxygen-free. All the experiments were conducted in triplicate to obtain the average and standard deviation of bio-oil yield.
The vapor-phase pyrolysis product was quenched using a condenser (RW1025G, JEIO TECH). The condensed liquid product (bio-oil) was analyzed using gas chromatography/mass spectrometry (GC/MS) (QP2010, Shimadzu, Japan) equipped with an HP-5 MS (30 m × 0.25 mm × 0.25 μm) capillary column. Bio-oil was dissolved in HPLC-grade acetone with a 1:10 mass ratio prior to analysis. After being stabilized for 5 min at 40 °C, temperature was increased to 200 °C at a rate of 5 °C/min and then to 300 °C at a rate of 20 °C/min. Then 300 °C was maintained for 10 min. The peaks appearing in the mass spectra obtained were interpreted using the NIST05 library.
The gas product that was not condensed in the condenser was collected in a Teflon bag for 20 min and was analyzed later using gas chromatography (ACME 6000, Young Lin Instrument, Co., Ltd., Anyang, Korea) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID). CO and CO2 were analyzed using TCD with a Carboxen 1000 (15 ft × 1/8 in) column, whereas FID with HP-plot Al2O3/KCl (50 m × 0.322 mm × 8.0 μm) column was used for the analysis of C1–C10 hydrocarbons.
3. Results and Discussion
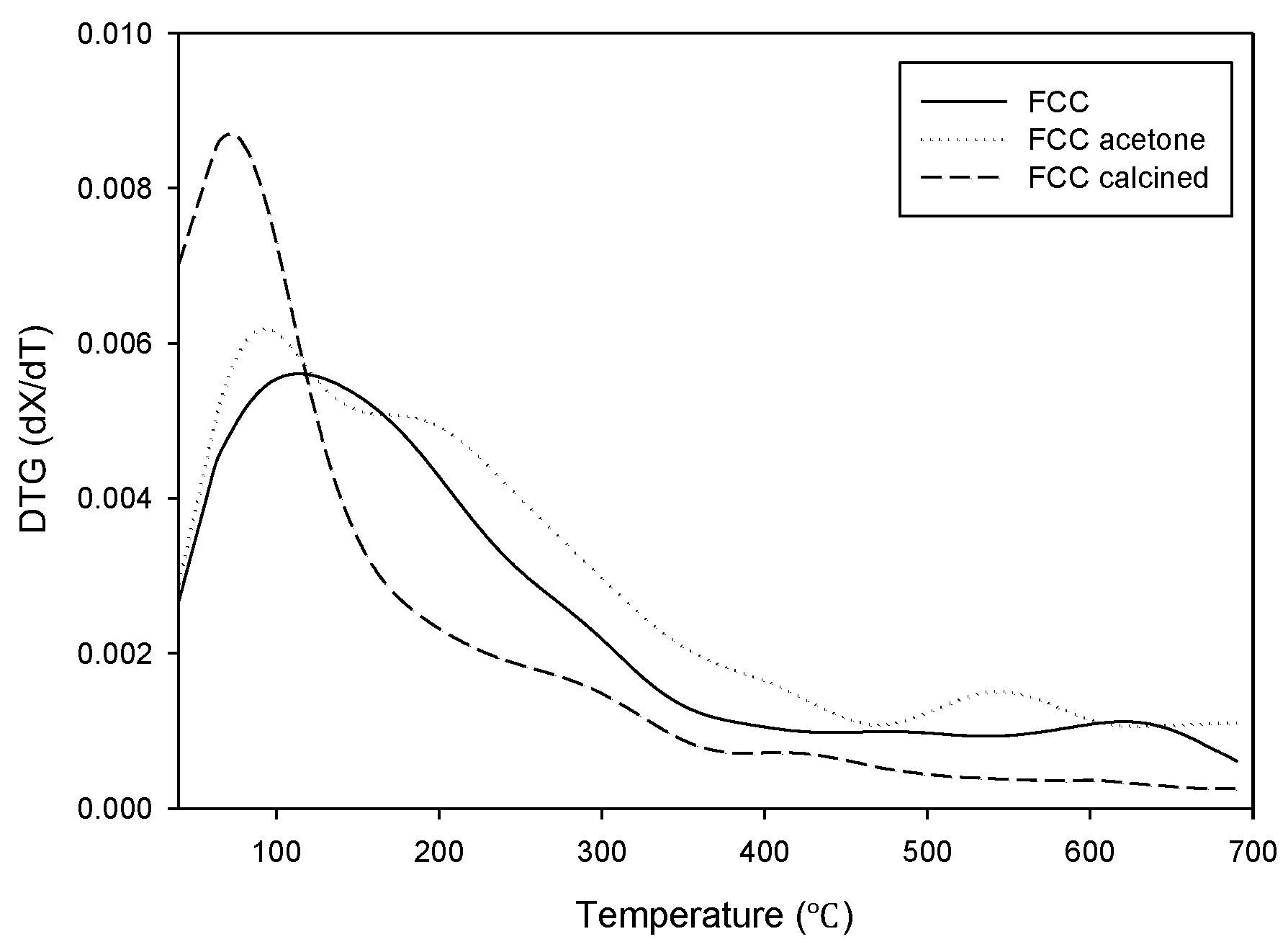
The TGA analysis results of the catalysts are shown in
Figure 1. The weight reduction is explained by the decomposition of carbonaceous material deposited on the surface of the waste catalyst. The mass of non-treated waste FCC catalyst (FCC) and the acetone-treated waste FCC catalyst (FCC-acetone) kept decreasing until the end of the analysis. FCC-acetone showed an even higher mass reduction than non-treated FCC during the TGA analysis, indicating that the physical activation was not very effective in removing the carbon deposit and that it rather caused the adsorption of acetone, leading to an increase of organic residue on the catalyst surface. On the other hand, the mass of chemically activated waste FCC catalyst (FCC-calcined) stopped decreasing at 550 °C, indicating that calcination was effective in removing organic deposit on the catalyst surface to a considerable extent. The derivative thermogravimetry (DTG) curves are shown in
Figure 2. Although all the three curves showed similar trend, FCC-calcined showed the highest maximum mass reduction rate around 100 °C.
The BET surface area, pore size, and pore volume of the catalysts were summarized in
Table 1. The specific surface area was increased significantly by both activation treatments but the effect of calcination was larger than that of acetone washing. Pore size and pore volume did not show large difference among the catalysts. Based on the TGA analysis and BET surface area measurement results, chemical activation was more effective in removing carbon deposit and organic residues on the catalyst surface. The XRD analysis showed that the waste FCC catalyst had the same basic structure as HY zeolite and that physical and chemical activations did not alter the crystalline structure of the catalyst.
Figure 3 shows the bio-oil yields obtained from the pyrolysis of reed conducted under different catalytic conditions. The error bar indicates the standard deviation obtained from the triplicate experiments. In the non-catalytic pyrolysis, the yields of oil, solid residue and gas were 50.7%, 22.0%, and 27.3%, respectively. Catalytic pyrolysis with a biomass-to-catalyst mass ratio of 1:1 led to a lower oil yield (and a higher gas yield) compared to non-catalytic pyrolysis mainly due to decomposition of oil species into gaseous species (with lower molecular mass). However, when the catalyst dosage was increased further (to a biomass-to-catalyst mass ratio of 1:10), the oil yield increased because the coke (solid residue) formation was suppressed by catalysts.
The GC-MS chromatogram of the bio-oil produced in this study showed more than 100 peaks. Therefore, all the bio-oil species detected in this study were grouped into seven categories: acids, oxygenates, phenolics, aliphatic hydrocarbons (aliphatics hereafter), monocyclic aromatic hydrocarbons (aromatics hereafter), polycyclic aromatic hydrocarbons (PAHs), and nitrogen-containing species (N compounds hereafter).
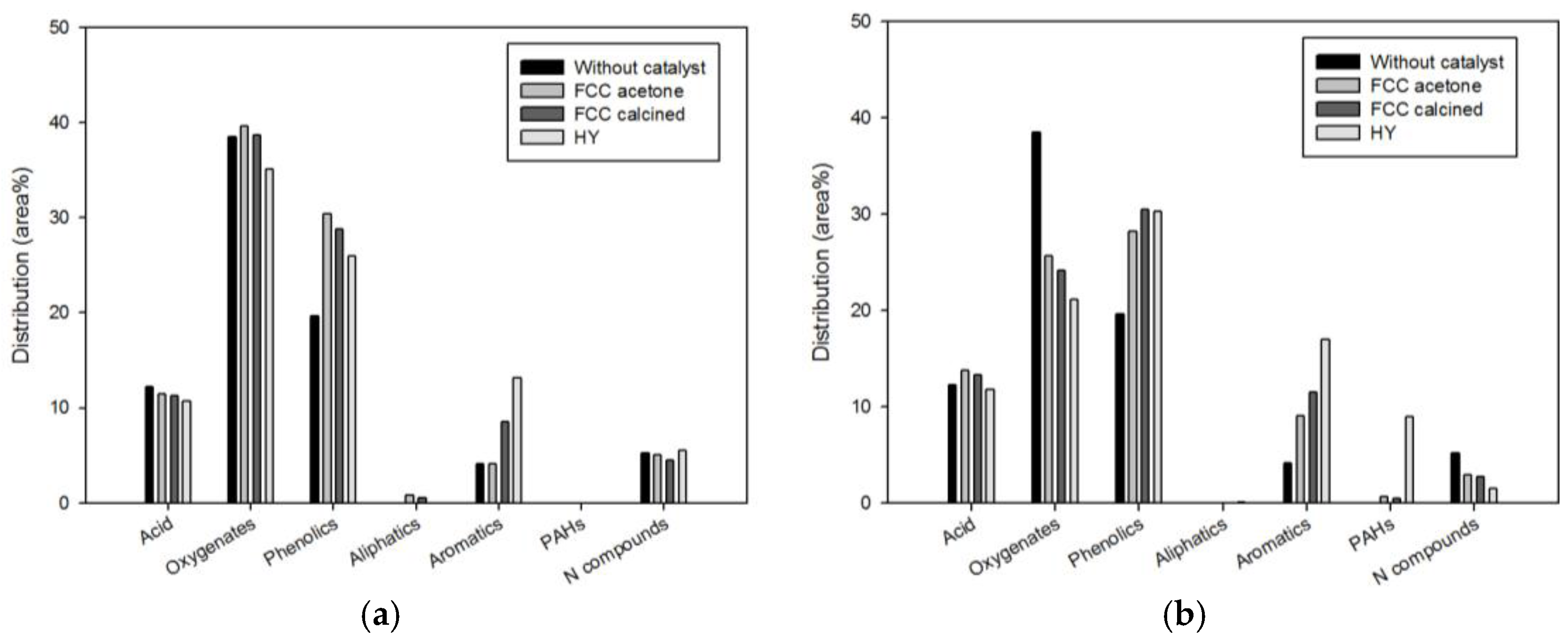
Figure 4 compares the bio-oil compositions (expressed in area %) obtained using different catalysts: acetone-washed waste FCC catalyst (FCC-acetone), waste FCC catalyst calcined for 3 h after acetone-washing (FCC-calcined), and commercial HY. The results obtained with two different biomass-to-catalyst ratios 1:1 and 1:10 are shown separately.
When the biomass-to-catalyst ratio was 1:1, phenolics (FCC-acetone, FCC-calcined, and HY) and aromatics (FCC-calcined and HY) were increased significantly by catalysis, while the contents of the other species were little affected by catalysis. When the biomass-to-catalyst ratio was 1:10, the content of oxygenates was reduced considerably, while the contents of phenolics and aromatics were increased significantly for all catalysts. The N compounds also decreased by all catalysts. Considerable increase in PAHs content was obtained only for HY. The conversion of oxygenates to aromatics over acid sites of zeolite-based catalysts is a well-known phenomenon; xylan, which is a thermally unstable fibrin linking cellulose and lignin, is easily decomposed on the acid sites and converted to aromatics through decarbonylation and aromatization [
13,
14].
Acids were mostly acetic acid. The content of acids did not change significantly as a result of catalysis regardless of the catalyst dosage. Acetic acid contained in liquid fuels can cause corrosion of pipes and engines, while it can be used for producing bulk-chemical, vinegar, cleansing agent, vinyl acetate, acetic anhydride, esters, solvent, and road deicer (as calcium acetate) if it is separated properly [
15]. Therefore, it must be separated from bio-oil before using the oil.
The main species among PAHs was naphthalene. Naphthalene is regarded as a high-value-added chemical because it can be widely used as a raw material for producing a number of chemical products, such as synthetic resin, pesticide and dye [
7]. PAHs were not produced from non-catalytic pyrolysis and from catalytic pyrolysis with the biomass-to-catalyst ratio of 1:1. On the other hand, when the biomass-to-catalyst ratio was 1:10, significant quantity (9.01%) of PAHs were detected for HY. The PAHs content obtained from waste FCC catalyst was marginal.
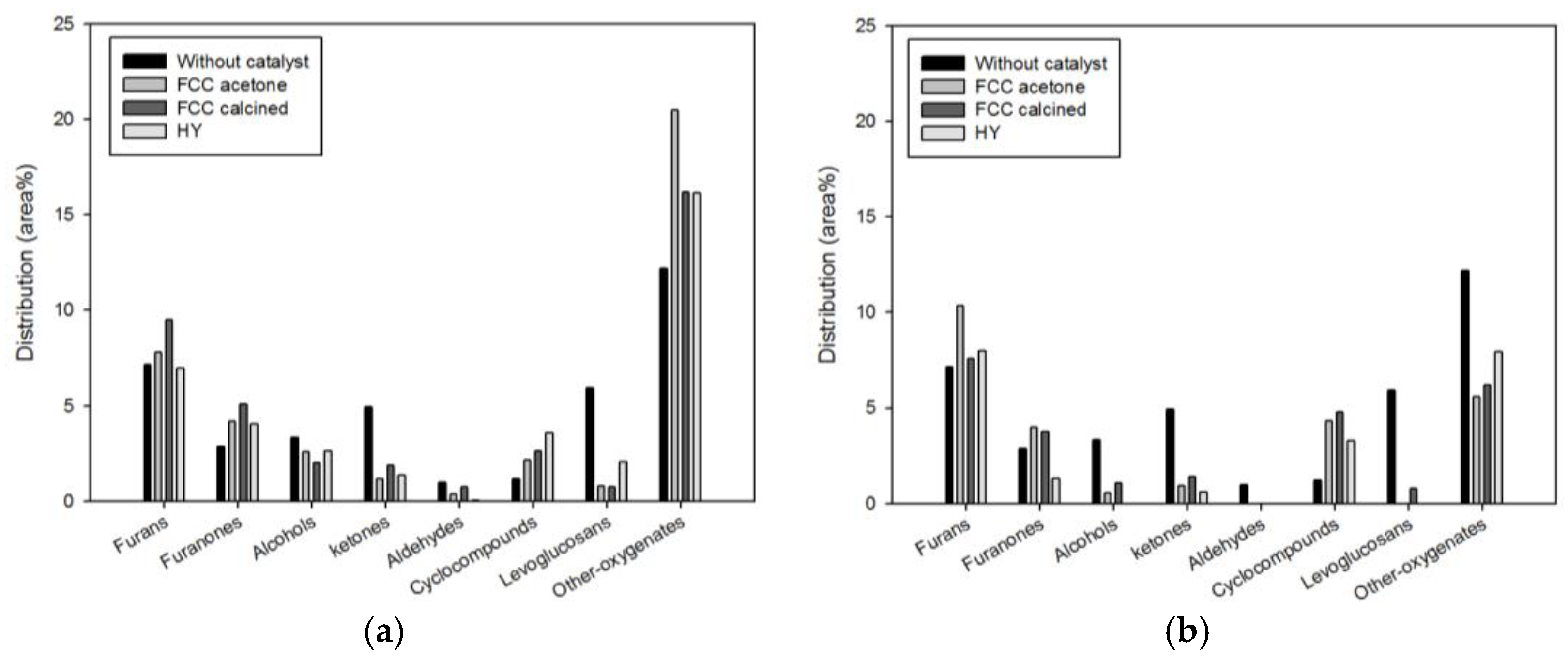
Figure 5 shows the detailed species distribution of oxygenates obtained under different catalytic conditions. While considerable quantity of levoglucosan (5.92%) was produced from non-catalytic pyrolysis, pyrolysis with the biomass-to-catalyst ratio of 1:1 reduced it dramatically: 0.78% (FCC-acetone), 0.75% (FCC-calcined), and 2.08% (HY). Further, when the biomass-to-catalyst ratio was 1:10, virtually no levoglucosan remained. The conversion of levoglucosan on the acid sites of catalysts to furans and aromatics has been reported repeatedly in previous studies [
6,
16,
17].
Furans are widely used in the petrochemical industry for producing furan resin, fuel additives, and medicines. Furans are known to be produced from the dehydration reaction of carbohydrate (e.g., levoglucosan) on the acid sites of catalysts [
16]. The increase in furans content due to catalysis was smaller than the reduction in levoglucosan content (
Figure 5). This was attributed to the conversion of levoglucosan to furanones and cyclocompounds due to dehydrogenation [
17] and the production of aromatics due to aromatization reaction [
16].
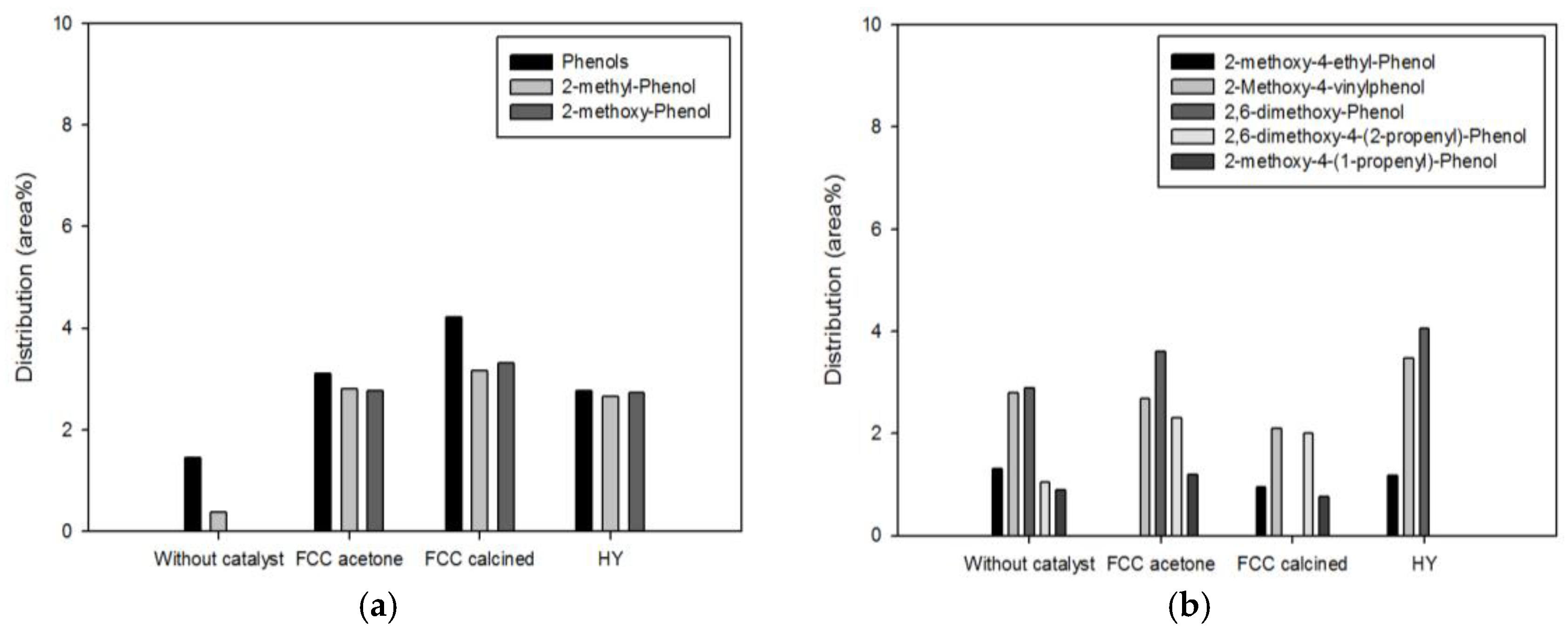
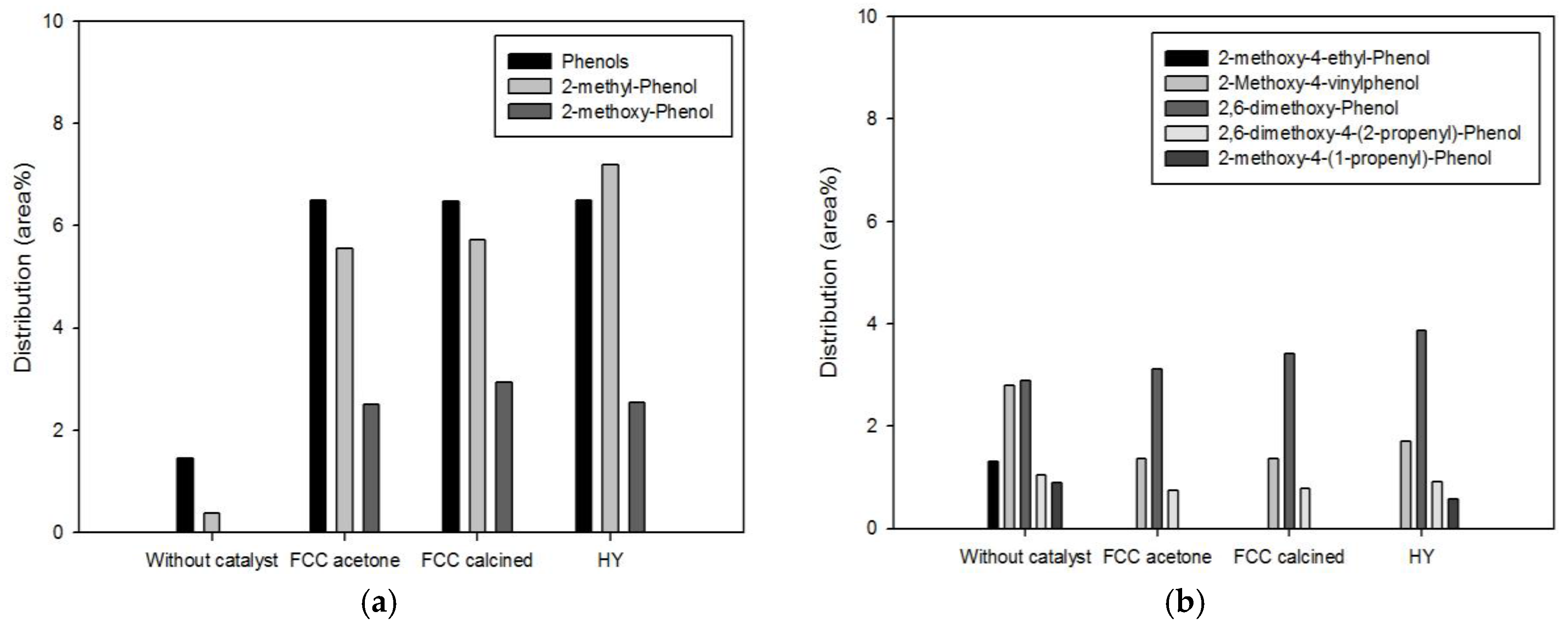
Phenolics are another high-value-added products of pyrolysis of biomass. They are produced from the decomposition of lignin contained in biomass.
Figure 6 and
Figure 7 show the detailed species distribution of phenolics obtained from different catalytic conditions. In both figures, low-molecular-mass phenolics (a) and high-molecular-mass phenolics (b) are shown separately. Pyrolysis with the biomass-to-catalyst ratio of 1:1 (
Figure 6) promoted the production of low-molecular-mass phenolics to a large extent, while high-molecular-mass phenolics were increased or decreased depending on the species. This result is attributed to promoted decomposition of lignin to phenolics and of high-molecular-mass phenolics to low-molecular-mass phenolics or aromatics owing to deoxygenation (dehydration, decarbonylation, and decarboxylation) and cracking occurring on the acid sites of the catalysts [
7,
18]. When the catalyst dosage was increased further (
Figure 7), the conversion of high-molecular-mass phenolics to low-molecular-mass phenolics was more evident. All the high-molecular-mass phenolics except 2,6-dimethoxy-phenol, whose molecular mass is the smallest among them, were decreased by pyrolysis. In particular, 2-methoxy-4-(1-propenyl)-phenol and 2-methoxy-4-vinylphenol, which were removed or considerably decreased by pyrolysis with the biomass-to-catalyst ratio of 1:10, are unsaturated compounds containing C=C bonds that are not inside benzene rings. Since they can cause the instability of fuels, the removal of these compounds is advantageous in terms of the oil quality improvement.
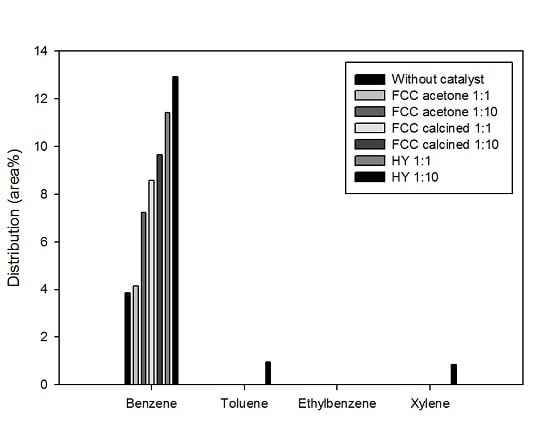
Figure 8 shows the most valuable four monocyclic aromatic compounds produced under different catalytic conditions: benzene, toluene, ethylbenzene, and xylene (often called BTEX). It is shown that most aromatics produced in this study were benzene; a little toluene and xylene were produced only by using HY with the biomass-to-catalyst ratio of 1:10. The production of aromatics was promoted by catalysis and the effect of catalysis was stronger when the catalyst dosage was higher. This result is in good agreement with the reports of previous studies that acid sites are required to produce aromatics during biomass pyrolysis [
4,
5,
7,
9,
16]. BTEX are very important raw materials in the petrochemical industry. Therefore, the production of BTEX using waste biomass and waste catalyst achieved in this study has deep implications for developing resources that can replace fossil fuels.
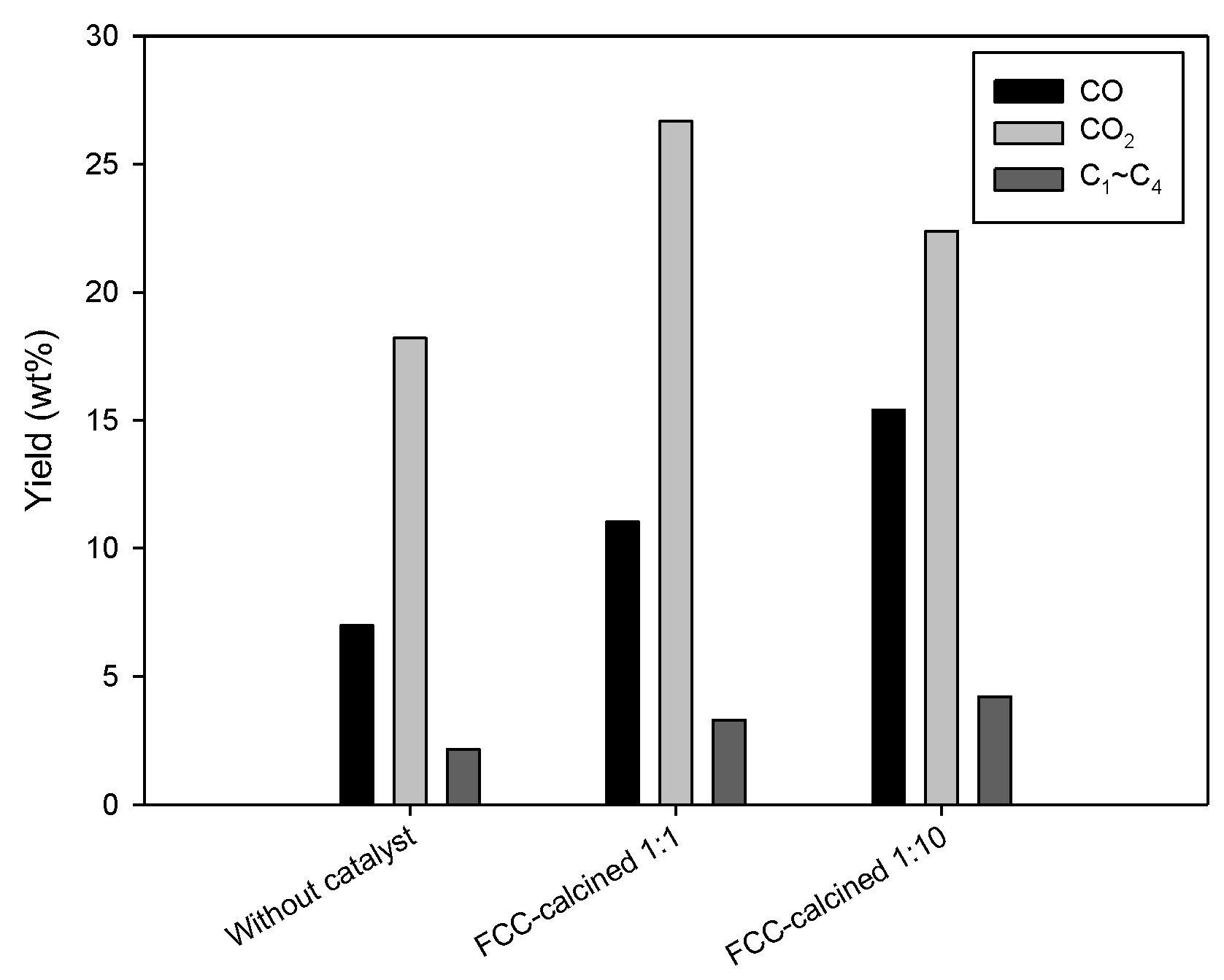
The gas products obtained over FCC-calcined with the biomass-to-catalyst ratio of 1:1 and 1:10 are shown in
Figure 9. The results obtained without catalyst are shown together. Catalysis apparently increased the production of C
1–C
4 hydrocarbon, CO, and CO
2. C
5–C
6 components were not detected, indicating that the condenser worked properly. Enhanced production of CO and CO
2 can be attributed to the catalytic deoxygenation (decarbonylation and decarboxylation) of oxygenates in bio-oil. Catalytic cracking of bio-oil components converted large-molecular-mass compounds into C
1–C
4 hydrocarbons. A high correlation between the enhanced CO production due to catalytically promoted decarbonylation and decarboxylation and increased production of aromatic hydrocarbons has been reported [
19], which is in good agreement with the result of this study. Increase in the catalyst dosage (change of the biomass-to-catalyst ratio from 1:1 to 1:10) resulted in reduced CO
2 production, while the production of C
1–C
4 hydrocarbons was enhanced. This can be attributed to enhanced catalytic demethylation and demethoxylation of lignin-derived phenolic compounds, resulting in reduced content of phenolics with methoxy functional groups in bio-oil [
20].













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}