
Deformation Modes and Anisotropy of Anti-Perovskite Ti3AN (A = Al, In and Tl) from First-Principle Calculations


Abstract
:1. Introduction
2. Calculations
2.1. Calculation Parameters
2.2. Structure Properties
3. Results and Discussion
3.1. Structure Properties
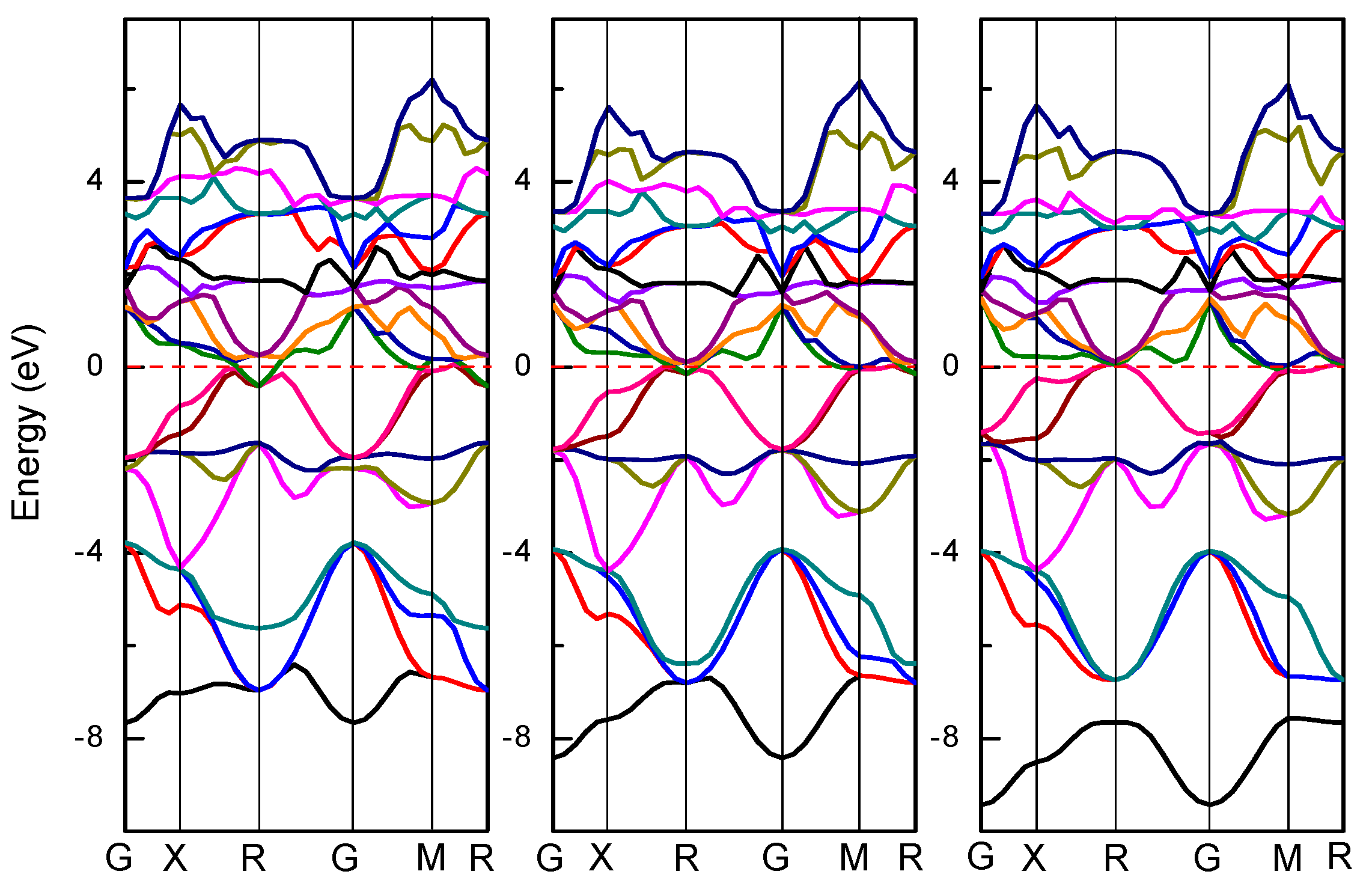
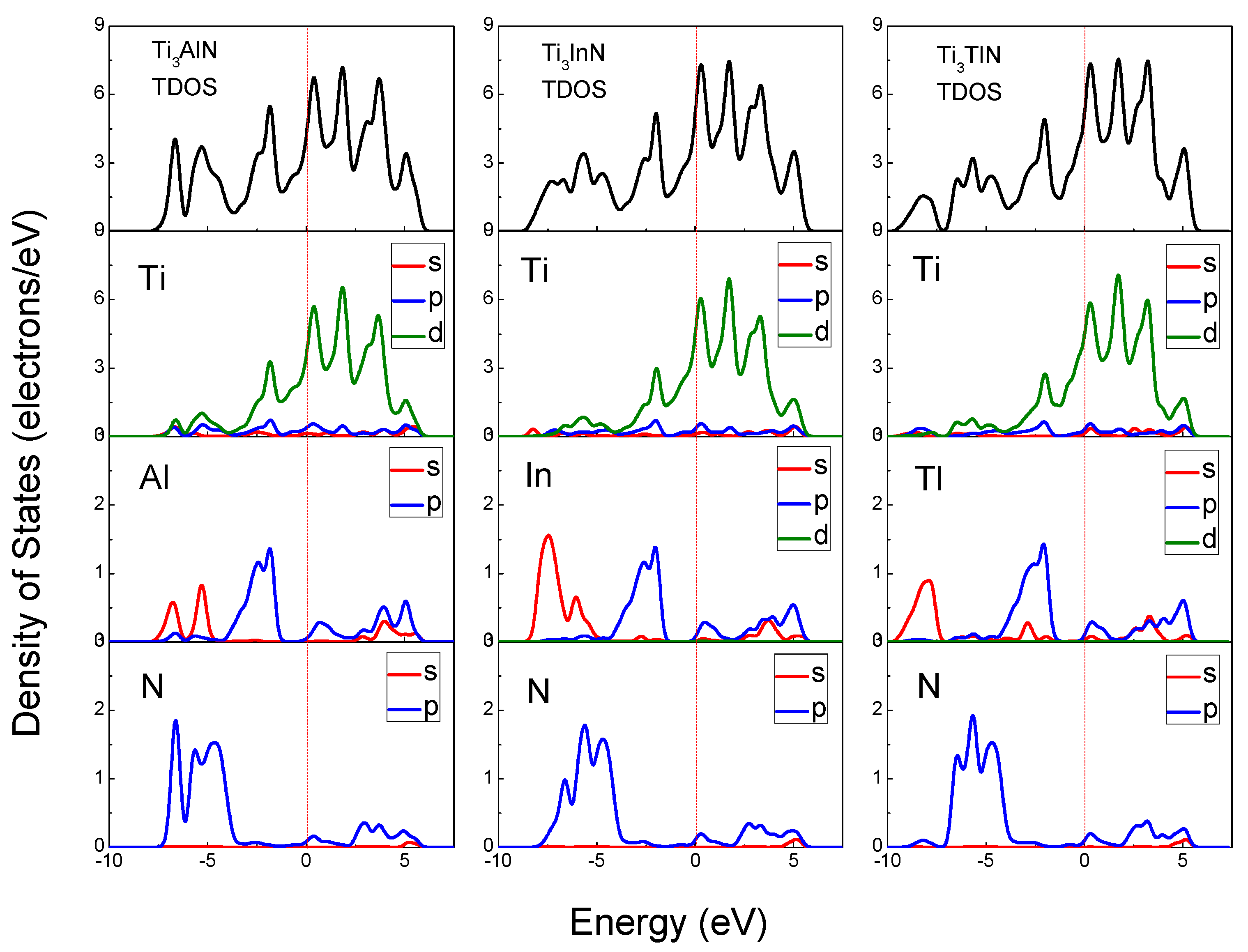
3.2. Electronic Structures
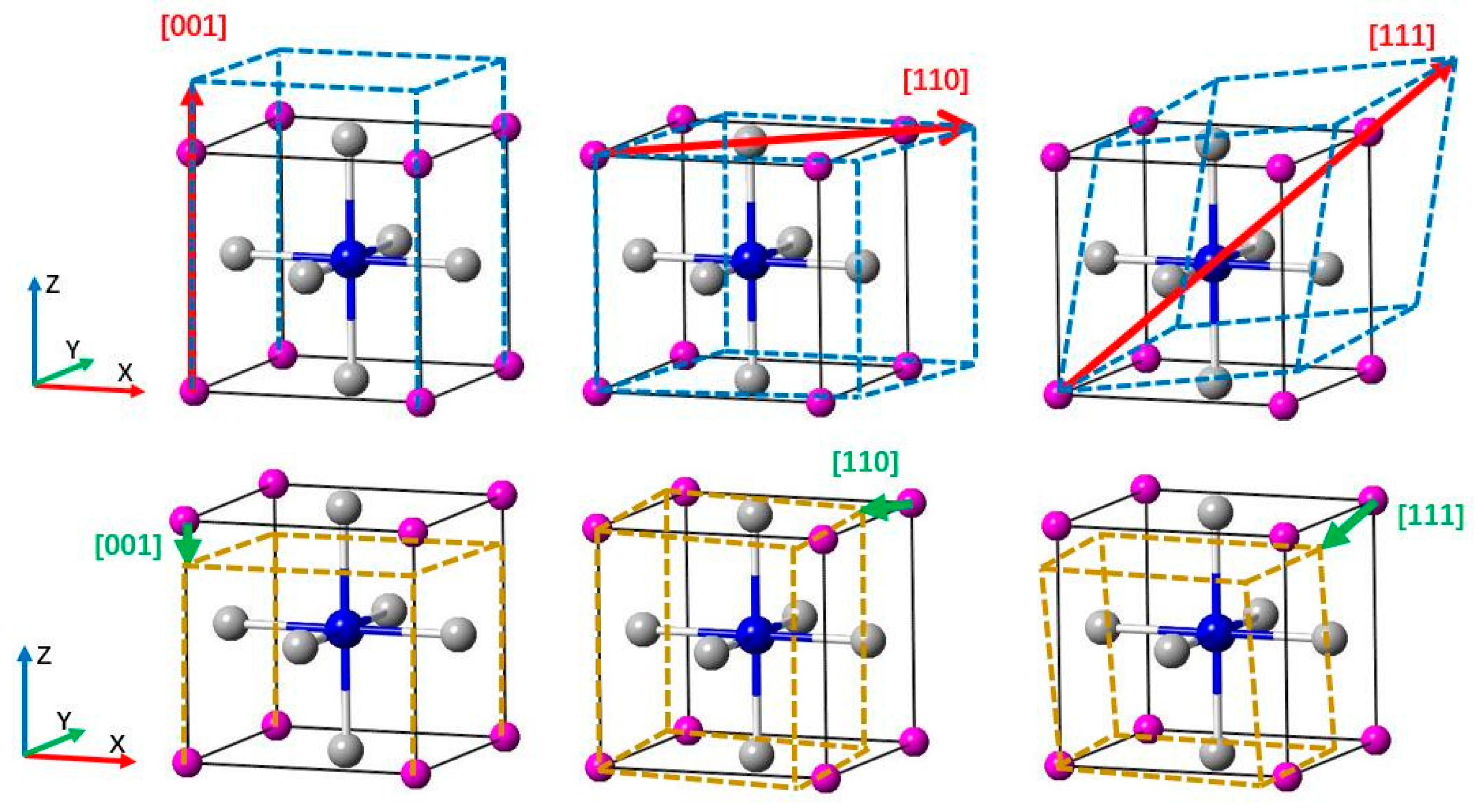
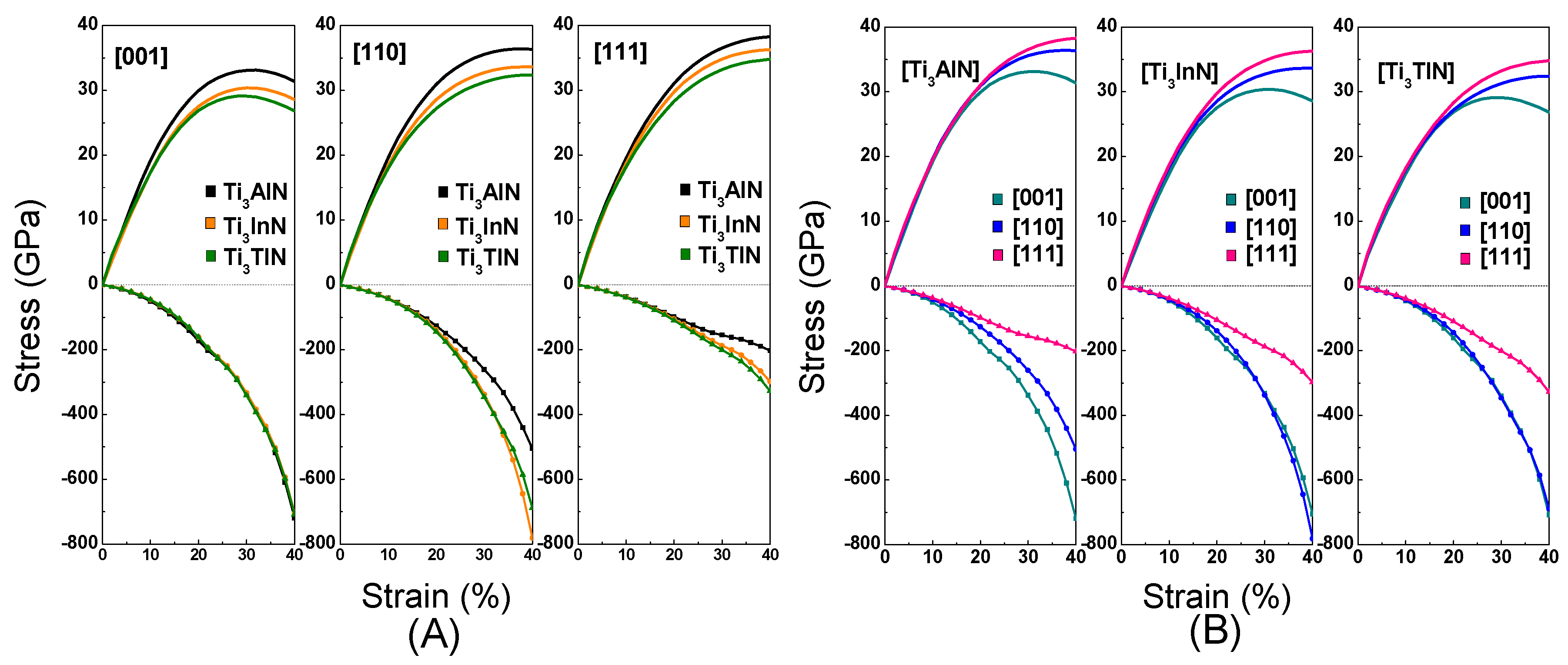
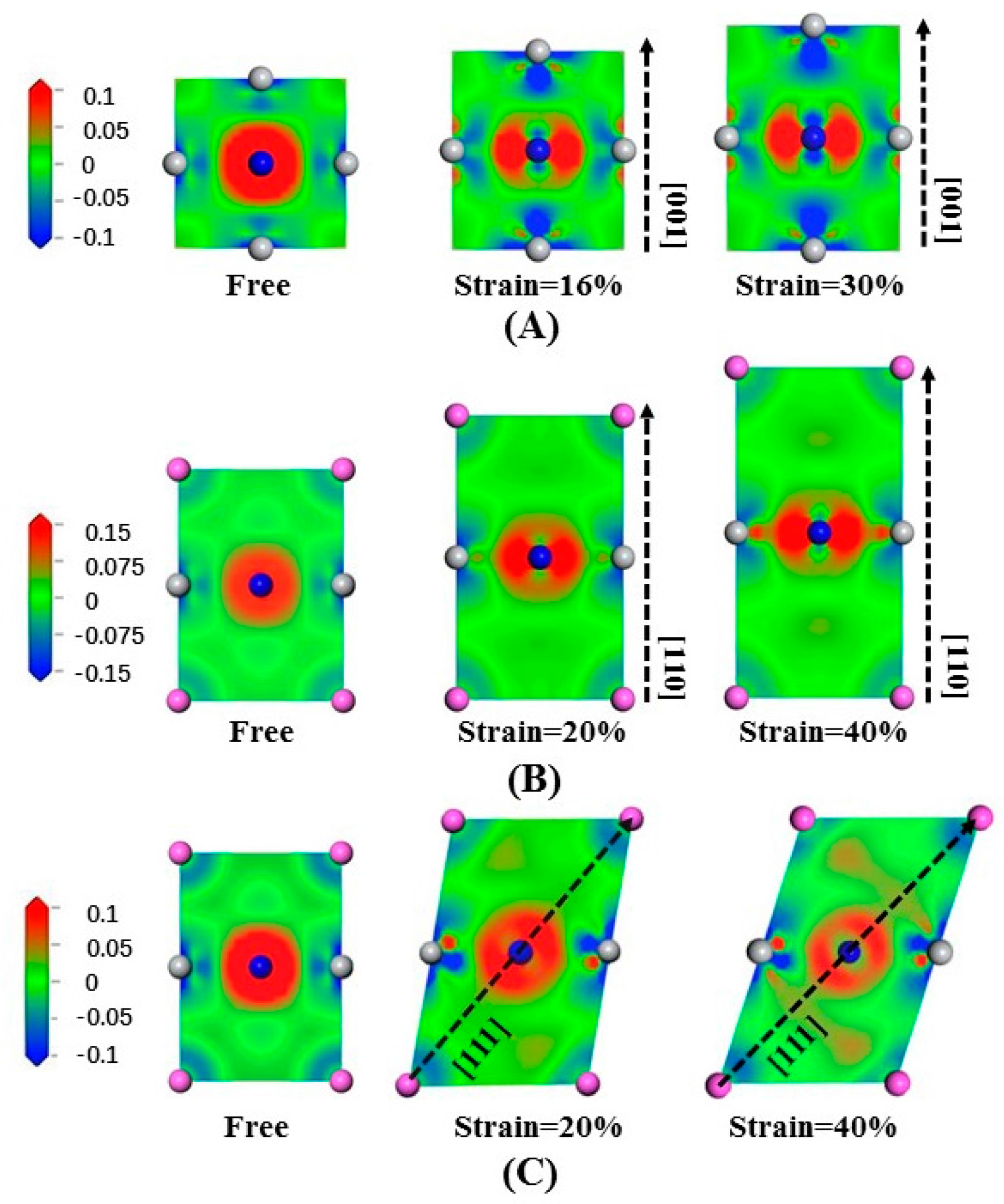
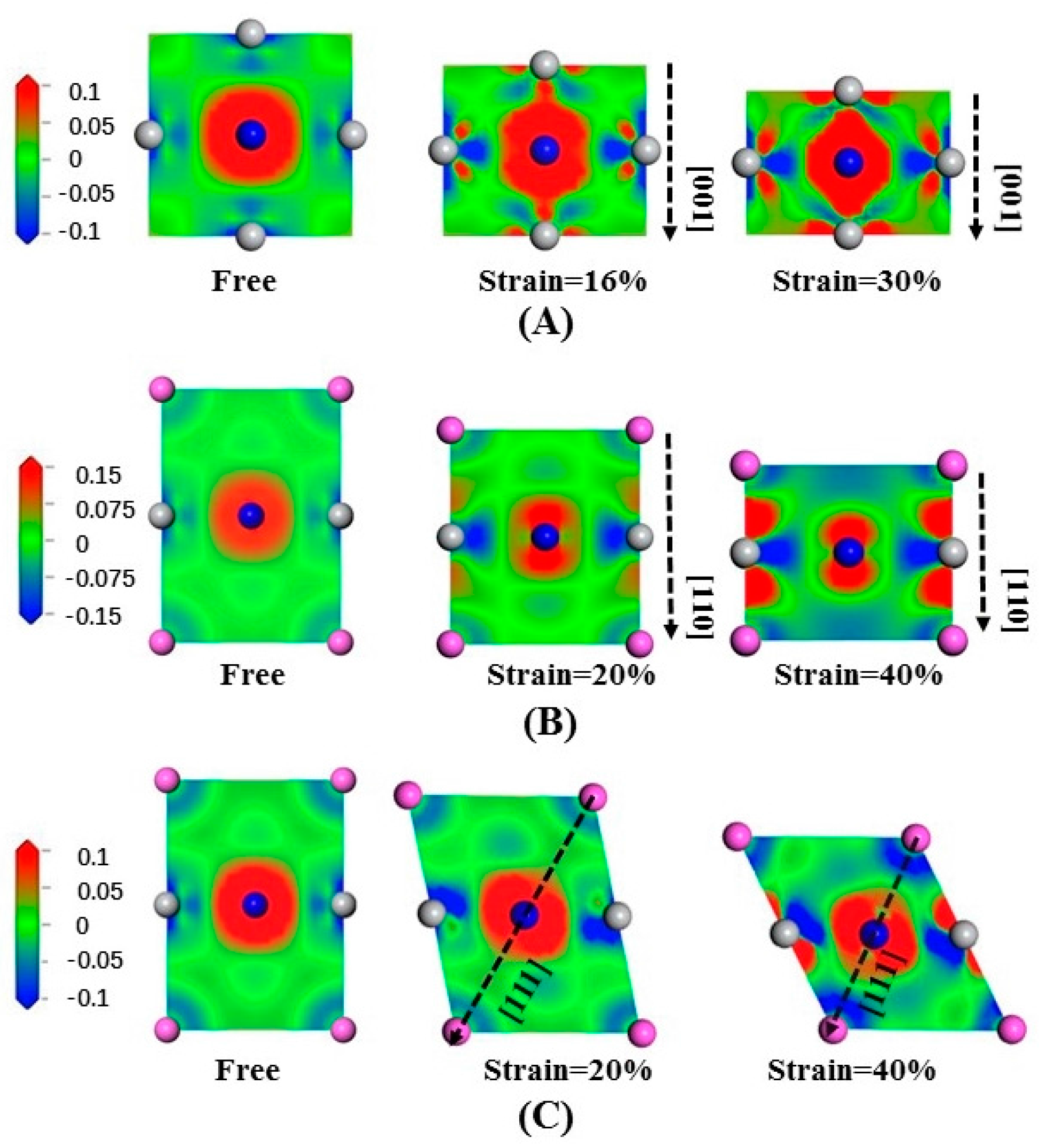
3.3. Deformation Modes
3.4. Hardness
3.5. Anisotropy of the Minimum Thermal Conductivity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dong, H.Y.; Yu, Y.D.; Jin, X.L. Microstructure and mechanical properties of SiC-SiC joints joined by spark plasma sintering. Ceram. Int. 2016, 42, 14463–14468. [Google Scholar] [CrossRef]
- Shrivastava, V. Microwave processed SrBi2Nb2O9 ferroelectric ceramics with controlled dielectric relaxation and metallic conduction. Ceram. Int. 2016, 42, 10122–10126. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Franke, P.; Seifert, H.J.; Wang, J.Y. Polymorphism of M3AlX Phases (M = Ti, Zr, Hf; X = C, N) and thermomechanical properties of Ti3AlN polymorphs. J. Am. Ceram. Soc. 2015, 98, 2570–2578. [Google Scholar]
- Nowotny, H.; Boller, H.; Beckmann, O. Alloy Phases Crystallizing with structures which occur with non-metallic compounds. Solid. State Chem. 1970, 2, 462–471. [Google Scholar] [CrossRef]
- Palmquist, J.P.; Li, S.; Persson, P.O.Å.; Emmerlich, J.; Wilhelmsson, O. Mn+1AXn phases in the Ti-Si-C system studied by thin-film synthesis and ab initio calculations. Phys. Rev. B 2004, 70, 165401. [Google Scholar] [CrossRef]
- Lin, Z.J.; Zhuo, M.J.; Zhou, Y.C.; Li, M.; Wang, J. Microstructures and Theoretical Bulk Modulus of Layered Ternary Tantalum Aluminum Carbides. J. Am. Ceram. Soc. 2006, 89, 3765–3769. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, B.; Wang, J.Y.; Zhou, Y.C. Low-temperature instability of Ti2SnC: A combined transmission electron microscopy, differential scanning calorimetry, and X-ray diffraction investigations. J. Mater. Res. 2009, 24, 39–49. [Google Scholar] [CrossRef]
- Schuster, J.C.; Bauer, J. The ternary system titanium-aluminum-nitrogen. J. Solid State Chem. 1984, 53, 260–265. [Google Scholar] [CrossRef]
- Tian, W.H.; Harada, K.; Nakashima, R.; Sano, T.; Nemoto, M. Crystal structures and morphologies of carbide and nitride precipitates in TiAl. J. Jpn. Inst. Met. 1993, 57, 1235–1243. [Google Scholar]
- Tan, J.; Han, H.; Wickramaratne, D.; Liu, W.; Zhao, M. A comparative first-principles study of the electronic, mechanical, defect and acoustic properties of Ti2AlC and Ti3AlC. J. Phys. D Appl. Phys. 2014, 47, 747–759. [Google Scholar] [CrossRef]
- Jäger, D.I.J.; Stahl, D.I.D.; Schmidt, P.C.; Kniep, R. Ca3AuN: A Calcium Auride Subnitride. Angew. Chem. Int. Ed. 1993, 32, 709–710. [Google Scholar] [CrossRef]
- Kim, W.S.; Chi, E.O.; Kim, J.C.; Choi, H.S.; Hur, N.H. Close correlation among lattice, spin, and charge in the manganese-based antiperovskite material. Solid State Commun. 2001, 119, 507–510. [Google Scholar] [CrossRef]
- Ivanovskii, A.L. Ternary carbides and nitrides based on transition metals and subgroup IIIB, IVB elements: Electronic structure and chemical bonding. Russ. Chem. Rev. 1996, 65, 461–478. [Google Scholar] [CrossRef]
- He, T.; Huang, Q.; Ramirez, A.P.; Wang, Y.; Regan, K.A.; Ragado, N. Superconductivity in the non-oxide perovskite MgCNi3. Nature 2001, 411, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Ming, Y.C.; Vennos, D.A.; Disalvo, F.J. Synthesis, structure, and properties of anti-perovskite nitrides Ca3MN, M = P, As, Sb, Bi, Ge, Sn, and Pb. J. Solid State Chem. 1992, 96, 415–425. [Google Scholar]
- Niewa, R.; Schnelle, W.; Wagner, F.R. Synthesis, Crystal Structure, and Physical Properties of (Ca3N)Tl. Allg. Chem. 2001, 627, 365–370. [Google Scholar] [CrossRef]
- Haddadi, K.; Bouhemadou, A.; Louail, L.; Rahal, F.; Maabed, S. Prediction study of the structural, elastic and electronic properties of ANSr3 (A = As, Sb and Bi). Comput. Mater. Sci. 2009, 46, 881–886. [Google Scholar] [CrossRef]
- Haddadi, K.; Bouhemadou, A.; Louail, L.; Medkour, Y. Structural, elastic and electronic properties of XNCa3 (X = Ge, Sn and Pb) compounds. Solid State Commun. 2009, 149, 619–624. [Google Scholar] [CrossRef]
- Uehara, M.; Yamazaki, T.; Kori, T.; Kashida, T.; Kimishima, Y.; Hase, I. Superconducting Properties of CdCNi3. J. Phys. Soc. Jpn. 2007, 76, 666–667. [Google Scholar]
- Vaitheeswaran, G.; Kanchana, V.; Svane, A.; Delin, A. Elastic properties of MgCNi3—A superconducting perovskite. J. Phys. Condens. Matter 2007, 19, 8568–8570. [Google Scholar] [CrossRef]
- Höglund, C.; Birch, J.; Beckers, M.; Alling, B.; Czigány, Z.S.; Mücklich, A. Sc3AlN—A New Perovskite. Eur. J. Inorg. Chem. 2008, 8, 1193–1195. [Google Scholar] [CrossRef]
- Kirchner, M.; Schinelle, W.; Wagner, F.R.; Niewa, R. Preparation, crystal structure and physical properties of ternary compounds (R3N)In, R = rare-earth metal. Solid State Sci. 2003, 5, 1247–1257. [Google Scholar] [CrossRef]
- Cherrad, D.; Selmani, L.; Maouche, D. First principles calculations on elasticity, electronic structure and bonding properties of antiperovskites ANTi3, (A = Al, In and Tl). J. Alloys Compd. 2011, 509, 4357–4362. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.L.D.; Probert, M.J.; Pickard, C.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Hohenberg, B.P.; Kohn, W. Inhomogeneous Electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B. 1976, 13, 5188. [Google Scholar] [CrossRef]
- Broyden, C.G. The convergence of a class of double rank minimization algorithms II. The new algorithm. J. Inst. Math. Appl. 1970, 6, 222–231. [Google Scholar] [CrossRef]
- Fletcher, R. Saturation and instability in acoustic paramagnetic resonance. Bull. Am. Meteorol. Soc. 1970, 3, 1349–1355. [Google Scholar]
- Goldfarb, D. A family of variable-metric methods derived by variational means. Math. Comp. 1970, 24, 23–26. [Google Scholar] [CrossRef]
- Shanno, D.F.; Kettler, P.C. Optimal Conditioning of Quasi-Newton Methods. Math. Comp. 1970, 24, 647–664. [Google Scholar] [CrossRef]
- Shim, J.H.; Kwon, S.K.; Min, B.I. Electronic structures of anti-perovskite superconductors MgXNi3 (X = B, C, and N). Phys. Rev. B 2001, 64, 607–611. [Google Scholar] [CrossRef]
- Voigt, W. Lehrbuch der Kristallphysik; B.G. Teubner: Leipzigz, Germany, 1928. [Google Scholar]
- Reuss, A.; Angew, Z. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. Math. Mech. 1929, 9, 49–58. (In Germen) [Google Scholar] [CrossRef]
- Güler, M.; Güler, E. Embedded Atom Method-Based Geometry Optimization Aspects of Body-Centered Cubic Metals. Chin. Phys. Lett. 2013, 30, 056201. [Google Scholar] [CrossRef]
- Jeitschko, W.; Nowotny, H.; Benesovsky, F. Die Kristallstruktur von Ti3InC, Ti3InN, Ti3TIC und Ti3TIN. Monatshefte für Chemie und verwandte Teile anderer Wissenschaften 1964, 95, 436–438. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823–843. [Google Scholar]
- Chen, Z.Q.; Peng, Y.S.; Hu, M.; Li, C.M.; Luo, Y.T. Elasticity, hardness, and thermal properties of ZrBn (n = 1, 2, 12). Ceram. Int. 2016, 42, 6624–6631. [Google Scholar] [CrossRef]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 1992, 8, 345–349. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Starzewski, M.O. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.H.; Buessem, W.R. The Elastic Anisotropy of Crystals. J. Appl. Phys. 1967, 38, 2010–2012. [Google Scholar] [CrossRef]
- Nye, F. Physical Properties of Crystals; Clarendon Press: Oxford, UK, 1964. [Google Scholar]
- Hearman, R.F.S. An Introduction to Applied Anisotropic Elasticity; Oxford University Press: Oxford, UK, 1961. [Google Scholar]
- Ono, S.; Kobayashi, M.; Tomoyose, T. Covalency of noble metal halides. Solid State Ion. 2005, 176, 363–366. [Google Scholar] [CrossRef]
- De Proft, F.; Van Alsenoy, C.; Peeters, A.; Langenaeker, W.; Geerlings, P. Atomic charges, dipole moments, and Fukui functions using the Hirshfeld partitioning of the electron density. J. Comput. Chem. 2002, 23, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Teter, D.M.; Hemley, R.J. Low-Compressibility Carbon Nitrides. Science 1996, 271, 53–55. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Seiler, P.; Dunitz, J.D.; Seiler, P. The absence of bonding electron density in certain covalent bonds as revealed by X-ray analysis. J. Am. Chem. Soc. 1983, 105, 7056–7058. [Google Scholar] [CrossRef]
- Gao, F.M.; Gao, L.H. Microscopic models of hardness. J. Superhard Mater. 2010, 32, 148–166. [Google Scholar] [CrossRef]
- Gou, H.Y.; Hou, L.; Zhang, J.W.; Gao, F.M. Pressure-induced incompressibility of ReC and effect of metallic bonding on its hardness. Appl. Phys. Lett. 2008, 92, 241901–241903. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Wan, C.L.; Qu, Z.X.; Huang, Z.C.; Chen, J.C.; Zhou, R.; Pan, W. Electronic structure, mechanical properties and thermal conductivity of Ln2Zr2O7 (Ln = La, Pr, Nd, Sm, Eu and Gd) pyrochlore. Acta Mater. 2011, 59, 1742–1760. [Google Scholar] [CrossRef]
- Zhang, X.W.; Wang, X.H.; Li, F.Z.; Zhou, Y.C. Mechanical and Thermal Properties of Antiperovskite Ti3AlC Prepared by an In Situ Reaction/Hot-Pressing Route. J. Am. Ceram. Soc. 2009, 92, 2698–2703. [Google Scholar] [CrossRef]
- Clarke, D.R. Materials selection guidelines for low thermal conductivity thermal barrier coatings. Surf. Coat. Technol. 2003, 163, 67–74. [Google Scholar] [CrossRef]
- Grimvall, G. Thermophysical Properties of Materials; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Cahill, D.G.; Watson, S.K.; Pohl, R.O. Lower limit to the thermal conductivity of disordered crystals. Phys. Rev. B 1992, 46, 6131–6140. [Google Scholar] [CrossRef]
- Vassen, R.; Cao, X.Q.; Tietz, F.; Basu, D.; Stöver, D. Zirconates as New Materials for Thermal Barrier Coatings. J. Am. Ceram. Soc. 2000, 83, 2023–2028. [Google Scholar] [CrossRef]
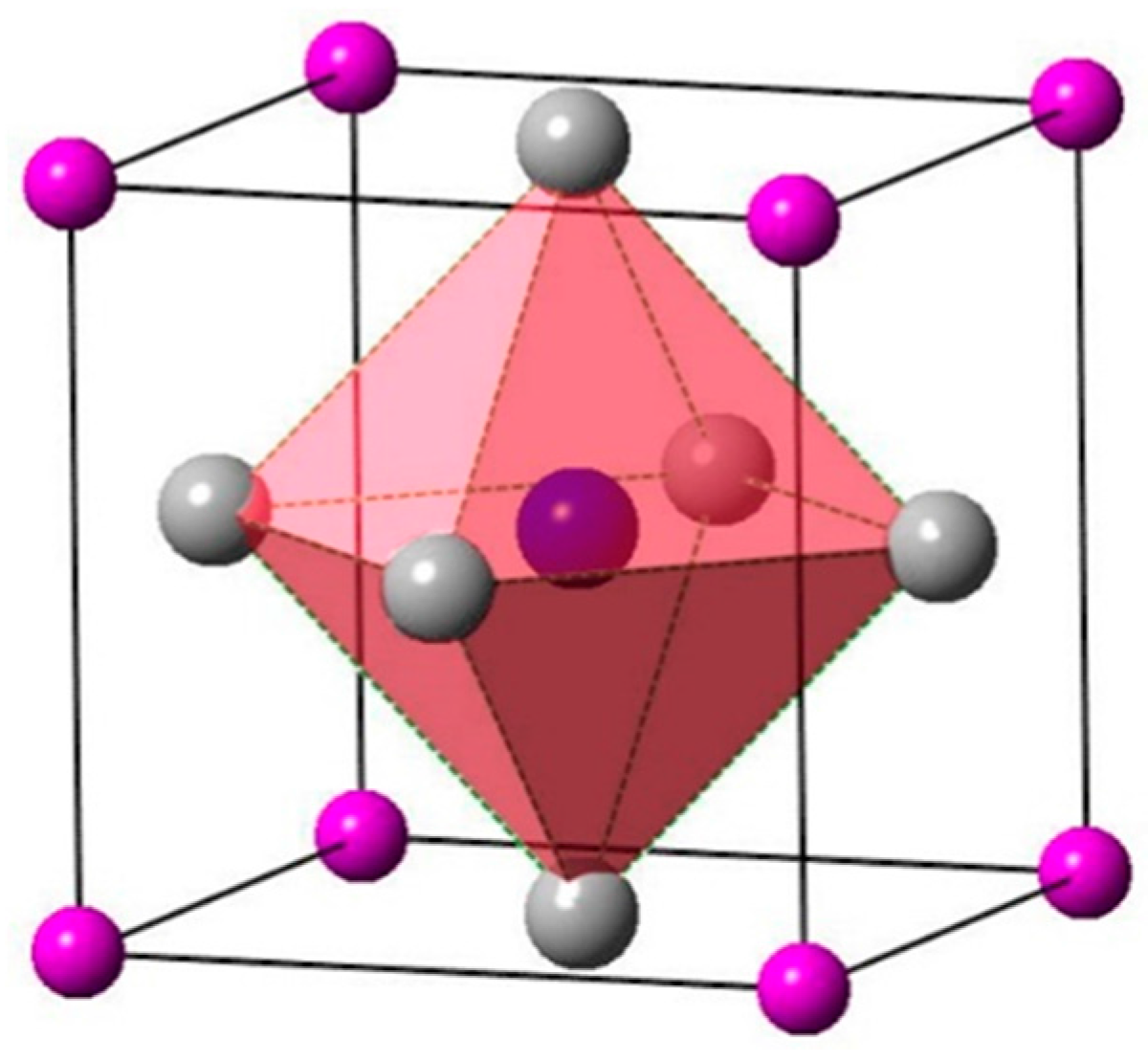
 : Ti atoms,
: Ti atoms,  : IIIA atoms,
: IIIA atoms,  : N atom (located in the body center).
: Ti atoms, : IIIA atoms, : N atom (located in the body center).
: N atom (located in the body center).
: Ti atoms, : IIIA atoms, : N atom (located in the body center).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Ti3AlN | Ti3InN | Ti3TlN | ||||||
|---|---|---|---|---|---|---|---|---|---|
| - | LDA | Ref. [23] | Expt. | LDA | Ref. [23] | Expt. | LDA | Ref. [23] | Expt. |
| a, b, c | 4.050 | 4.051 | 4.112 [36] | 4.114 | 4.116 | 4.190 [37] | 4.122 | 4.124 | 4.191 [37] |
| ρ | 4.636 | - | - | 6.525 | - | - | 8.619 | - | - |
| C11 | 239.07 | 239.94 | - | 196.48 | 189.85 | - | 258.97 | 256.36 | - |
| C12 | 159.09 | 156.60 | - | 172.69 | 171.05 | - | 145.25 | 138.44 | - |
| C44 | 57.63 | 57.91 | - | 45.62 | 49.68 | - | 62.38 | 63.76 | - |
| B | 185.75 | 184.38 | - | 180.62 | 177.32 | - | 183.16 | 177.75 | - |
| G | 49.78 | 50.76 | - | 26.75 | 25.93 | - | 60.11 | 61.79 | - |
| E | 137.10 | 139.47 | - | 76.48 | 74.19 | - | 162.56 | 166.13 | - |
| G/B | 0.27 | - | - | 0.15 | - | - | 0.33 | - | - |
| ν | 0.377 | - | - | 0.43 | - | - | 0.35 | - | - |
| B/C44 | 3.22 | - | - | 3.96 | - | - | 2.93 | - | - |
| C12-C44 | 101.46 | - | - | 127.07 | - | - | 82.87 | - | - |
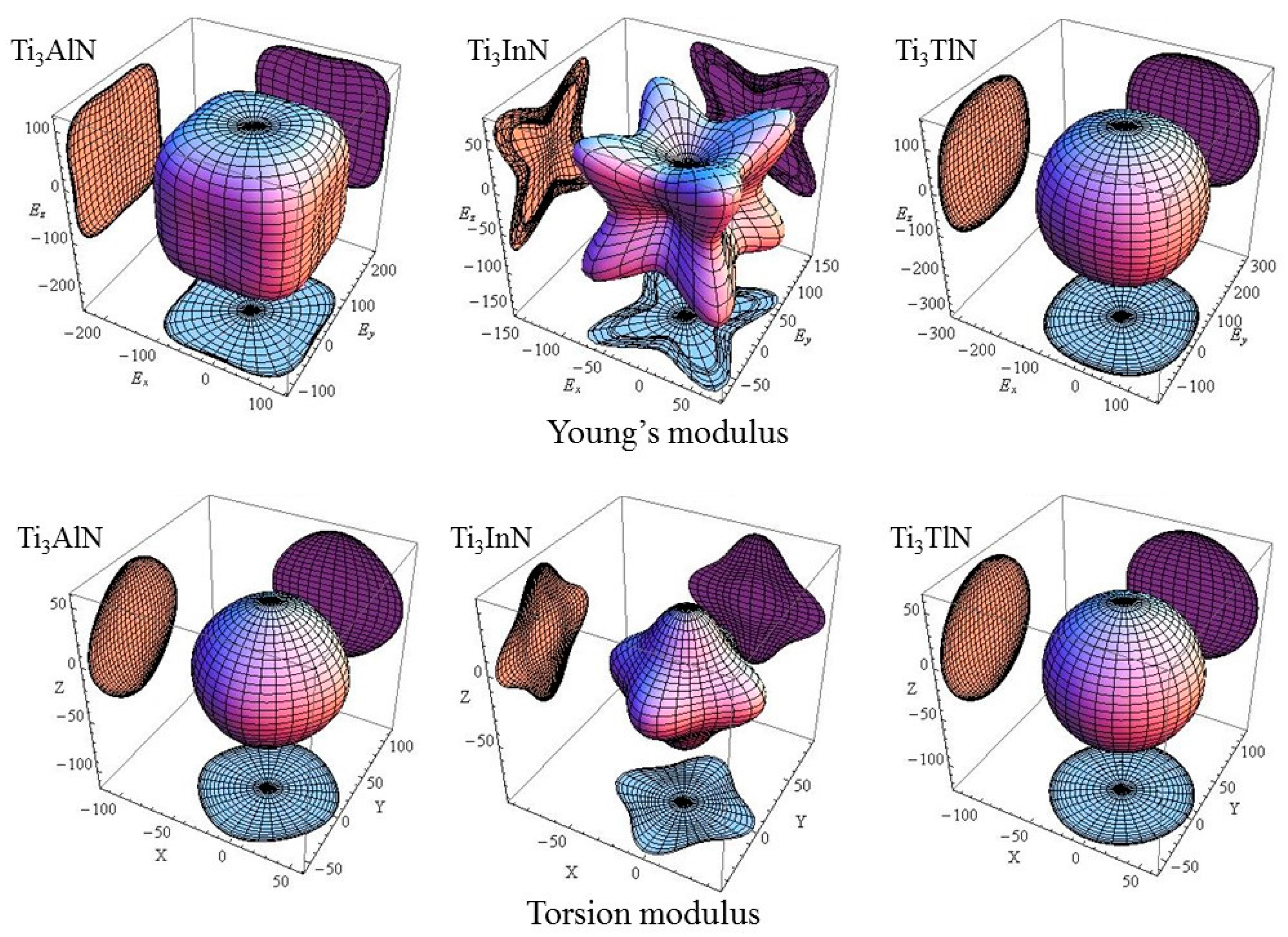
| Species | AU | AE | AG | AB | E[001] | E[110] | E[111] | T[100] | T[110] | T[111] |
|---|---|---|---|---|---|---|---|---|---|---|
| Ti3AlN | 0.162 | 0.015 | 0.016 | 0 | 111.9 | 142.5 | 152.9 | 57.6 | 54.6 | 53.9 |
| Ti3InN | 2.515 | 0.192 | 0.201 | 0 | 34.9 | 76.3 | 108.5 | 45.6 | 33.7 | 31.6 |
| Ti3TlN | 0.010 | 0.001 | 0.001 | 0 | 154.6 | 164.5 | 167.2 | 62.4 | 61.6 | 61.5 |
| Species | Bond | Nb | Length | Population | Vb | Hvb | Hv | |
|---|---|---|---|---|---|---|---|---|
| Ti3AlN | Ti-N | 3 | 2.025 | 0.51 | 5.78 | 20.26 | 10.73 | - |
| Ti-Al | 3 | 2.864 | 0.81 | 16.36 | 5.69 | |||
| Ti3InN | Ti-N | 3 | 2.057 | 0.60 | 6.06 | 22.03 | 6.87 | - |
| Ti-Ti | 3 | 2.909 | 0.33 | 17.15 | 2.14 | |||
| Ti3TlN | Ti-N | 3 | 2.061 | 0.61 | 6.10 | 22.18 | 11.14 | - |
| Ti-Ti | 3 | 2.915 | 0.87 | 17.25 | 5.59 | |||
| Ti3AlC | Ti-C | 3 | 2.052 | 0.62 | 6.02 | 17.02 | 11.27 | 7.8~12.5 [54] |
| Ti-Al | 3 | 2.902 | 0.84 | 23.04 | 5.52 |
| Species | [100] | [110] | [111] | ||||
|---|---|---|---|---|---|---|---|
| Ti3AlN | 3.28 | 7.38 | 2.94 | 3.53 | 7.44 | 3.15 | 7.53 |
| Ti3InN | 2.02 | 5.76 | 1.35 | 2.64 | 5.94 | 1.88 | 6.08 |
| Ti3TlN | 2.64 | 5.53 | 2.57 | 2.69 | 5.54 | 2.61 | 5.56 |
| CaOcal | 4.70 | 8.10 | 7.03 | 4.70 | 7.94 | 4.88 | 7.89 |
| CaOexp [56] | 4.94 | 8.21 | 7.02 | 4.94 | 8.19 | 4.96 | 8.18 |
| Species | Clark | Cahill | |||||
|---|---|---|---|---|---|---|---|
| Ma (10−23) | P × 1028 | ||||||
| Ti3AlN | 6.13 | 1.17 | 3.28 | 7.34 | 7.53 | 1.38 | - |
| Ti3InN | 9.05 | 0.71 | 2.02 | 5.76 | 7.18 | 0.94 | - |
| Ti3TlN | 12.03 | 0.90 | 2.64 | 5.51 | 7.14 | 1.04 | - |
| ZrO2 | 6.83 | 1.74 | 4.31 | 8.14 | 9.45 | 1.94 | 2.2 [58] |
| Species | [100] | [110] | [111] | (avg) |
|---|---|---|---|---|
| Ti3AlN | 1.384 | 1.381 | 1.373 | 1.379 |
| Ti3InN | 0.943 | 0.955 | 0.946 | 0.948 |
| Ti3TlN | 1.036 | 1.035 | 1.033 | 1.035 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.; Li, C.; Hu, M.; Hou, X.; Li, C.; Chen, Z. Deformation Modes and Anisotropy of Anti-Perovskite Ti3AN (A = Al, In and Tl) from First-Principle Calculations. Materials 2017, 10, 362. https://doi.org/10.3390/ma10040362
Chen K, Li C, Hu M, Hou X, Li C, Chen Z. Deformation Modes and Anisotropy of Anti-Perovskite Ti3AN (A = Al, In and Tl) from First-Principle Calculations. Materials. 2017; 10(4):362. https://doi.org/10.3390/ma10040362
Chicago/Turabian StyleChen, Kuankuan, Cong Li, Meng Hu, Xun Hou, Chunmei Li, and Zhiqian Chen. 2017. "Deformation Modes and Anisotropy of Anti-Perovskite Ti3AN (A = Al, In and Tl) from First-Principle Calculations" Materials 10, no. 4: 362. https://doi.org/10.3390/ma10040362
APA StyleChen, K., Li, C., Hu, M., Hou, X., Li, C., & Chen, Z. (2017). Deformation Modes and Anisotropy of Anti-Perovskite Ti3AN (A = Al, In and Tl) from First-Principle Calculations. Materials, 10(4), 362. https://doi.org/10.3390/ma10040362