
Generating Bulk-Scale Ordered Optical Materials Using Shear-Assembly in Viscoelastic Media


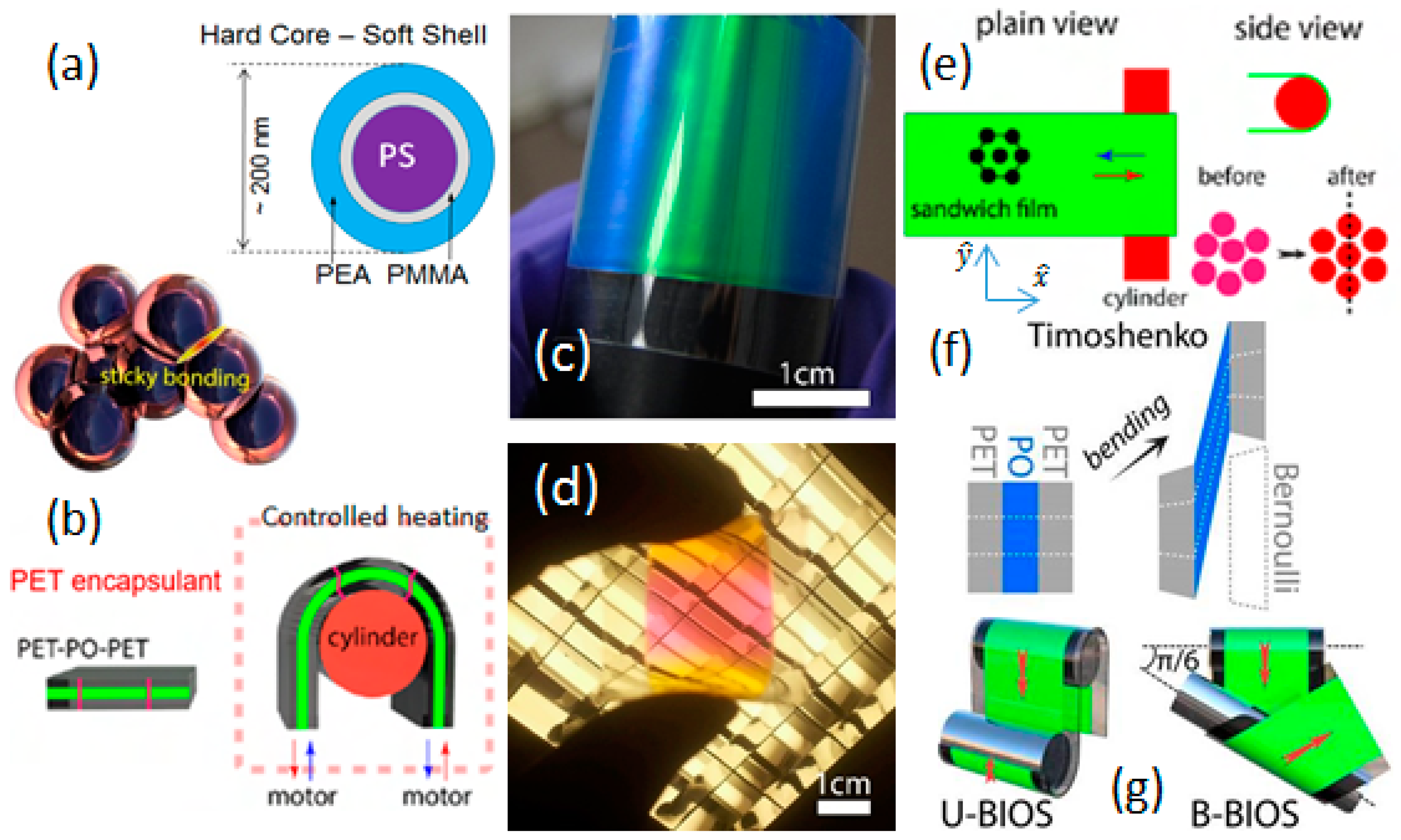{kind=link}
{kind=link}
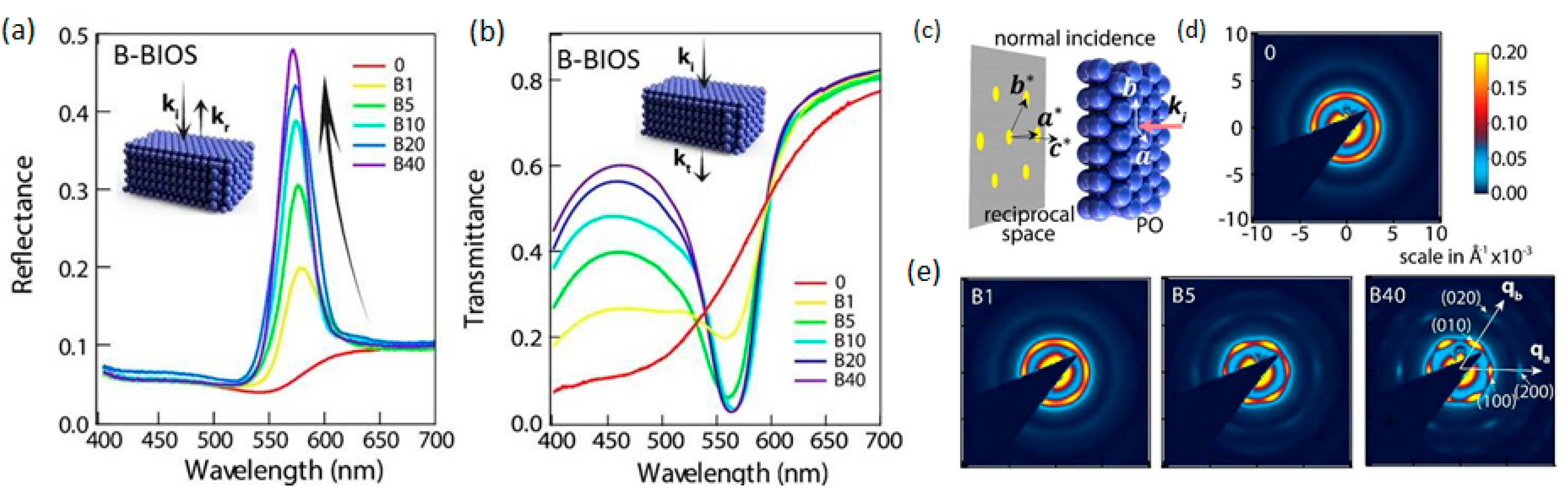{kind=link}
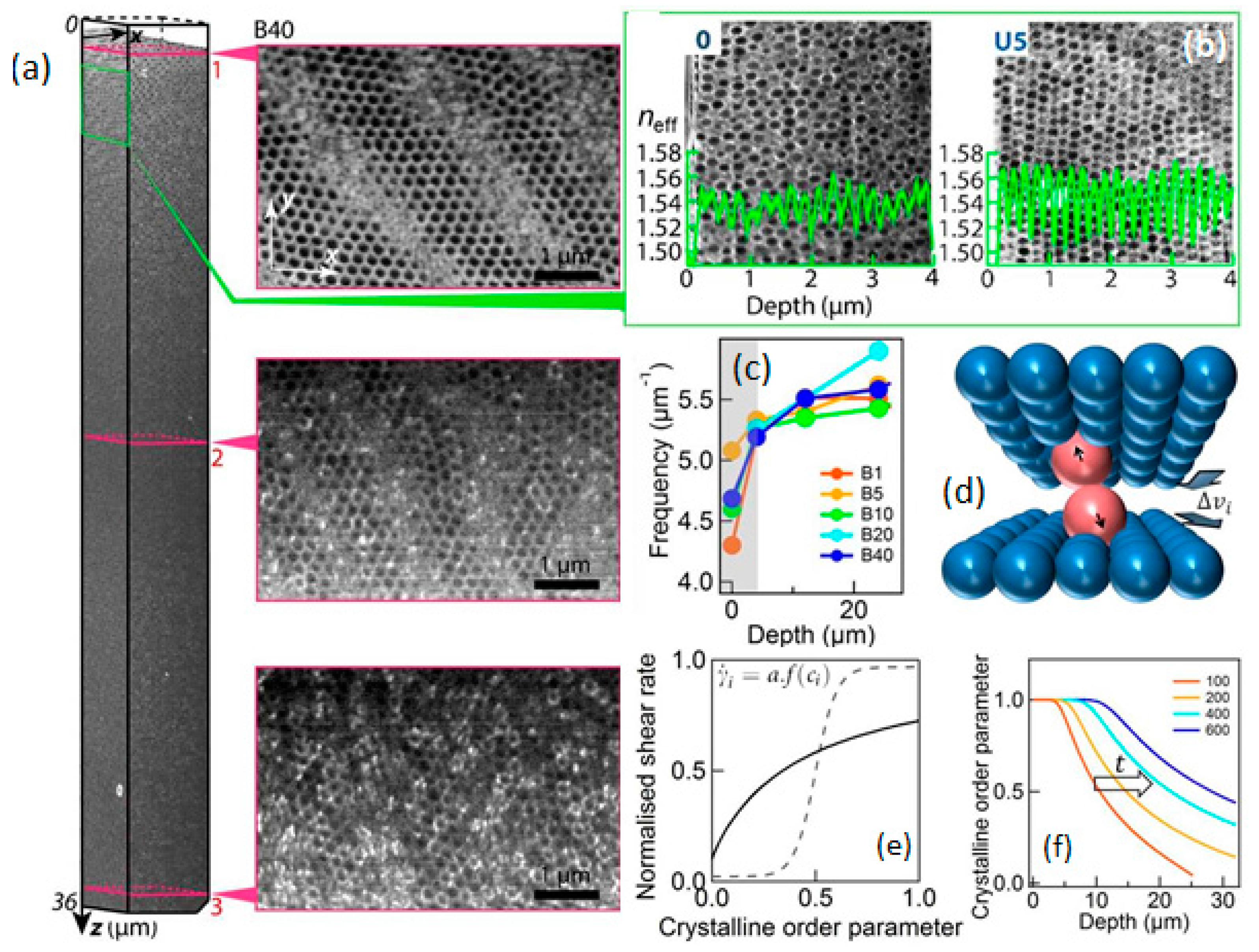{kind=link}
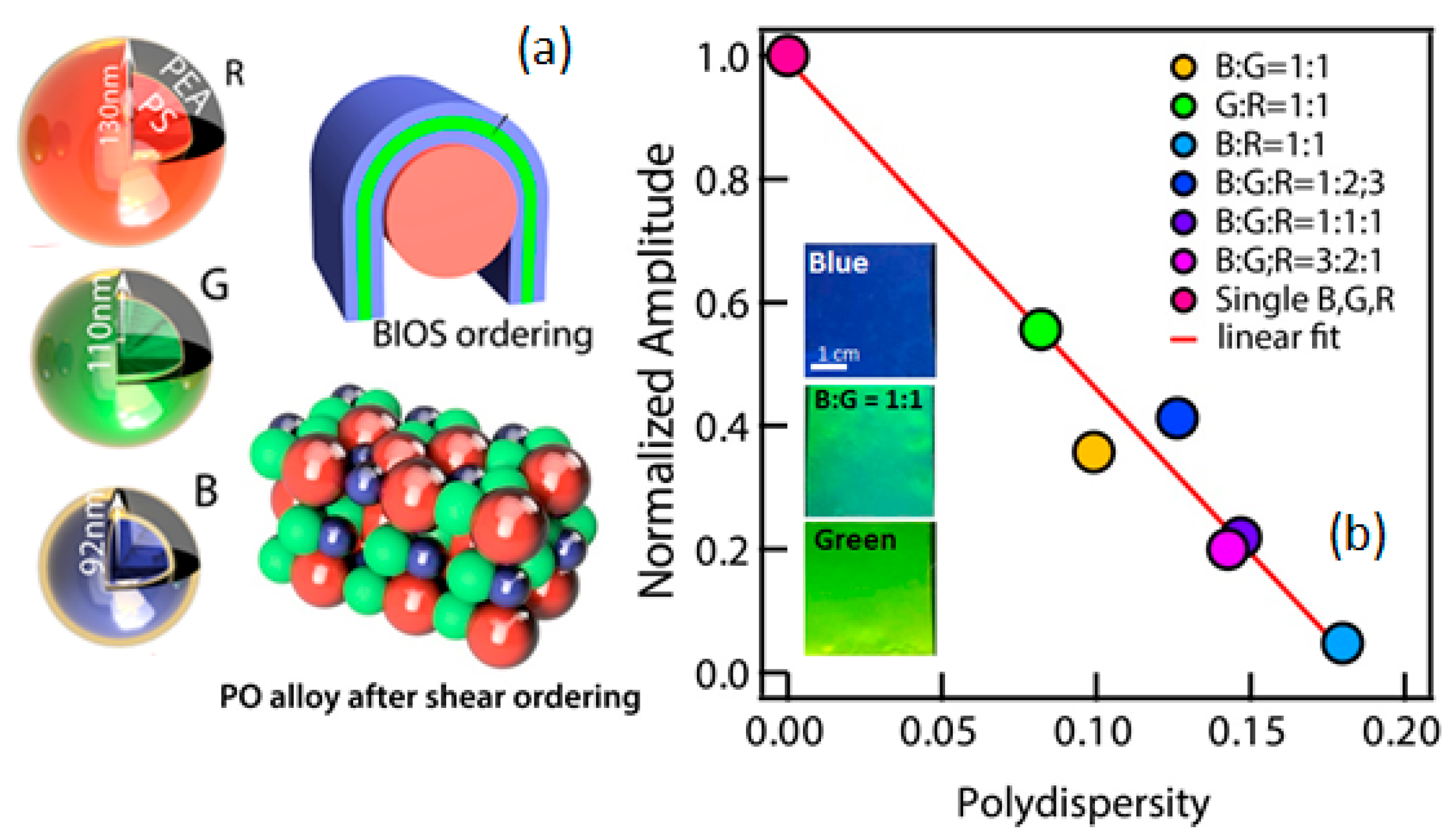{kind=link}
Abstract
:1. Introduction
1.1. Overview of Synthetic Opal Production
1.2. Shear-Ordering in Viscoelastic Media
2. Overview of Recent Studies
2.1. Processing Methods; Bending-Induced Oscillatory Shear
2.2. Rheometry and Time-Dependance of Shear Ordering
2.3. Optical Properties and Crystalline Ordering
2.4. Theoretical Framework
2.5. Nanoassembly of Polydisperse Particles; Polymer Opal “Alloys”
3. Discussion
4. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vukusic, P.; Sambles, J.; Lawrence, C. Structural colour—Colour mixing in wing scales of a butterfly. Nature 2000, 404, 457. [Google Scholar] [CrossRef] [PubMed]
- Vignolini, S.; Rudall, P.; Rowland, A.; Reed, A.; Moyroud, E.; Faden, R.; Baumberg, J.; Glover, B.; Steiner, U. Pointillist structural color in pollia fruit. Proc. Natl. Acad. Sci. USA 2012, 109, 15712–15715. [Google Scholar] [CrossRef] [PubMed]
- Marlow, F.; Muldarisnur; Sharifi, P.; Brinkmann, R.; Mendive, C. Opals: Status and prospects. Angew. Chem. Int. Ed. 2009, 48, 6212–6233. [Google Scholar] [CrossRef] [PubMed]
- Joannopoulos, J.D. Photonic Crystals; Princeton: Princeton, NJ, USA, 2008. [Google Scholar]
- Bermel, P.; Luo, C.; Zeng, L.; Kimerling, L.; Joannopoulos, J. Improving thin-film crystalline silicon solar cell efficiencies with photonic crystals. Opt. Express 2007, 15, 16986–17000. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Koh, C.; Singer, J.; Jeon, S.; Maldovan, M.; Stein, O.; Thomas, E. 25th anniversary article: Ordered polymer structures for the engineering of photons and phonons. Adv. Mater. 2014, 26, 532–568. [Google Scholar] [CrossRef] [PubMed]
- Knop, K. Diffraction gratings for color filtering in zero diffraction order. Appl. Opt. 1978, 17, 3598–3603. [Google Scholar] [CrossRef] [PubMed]
- Kolle, M.; Lethbridge, A.; Kreysing, M.; Baumberg, J.; Aizenberg, J.; Vukusic, P. Bio-inspired band-gap tunable elastic optical multilayer fibers. Adv. Mater. 2013, 25, 2239–2245. [Google Scholar] [CrossRef] [PubMed]
- Slesarenko, V.; Rudykh, S. Harnessing viscoelasticity and instabilities for tuning wavy patterns in soft layered composites. Soft Matter 2016, 12, 3677–3682. [Google Scholar] [CrossRef] [PubMed]
- De La Rue, R. Photonic crystals: Microassembly in 3d. Nat. Mater. 2003, 2, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Vlasov, Y.; Bo, X.; Sturm, J.; Norris, D. On-chip natural assembly of silicon photonic bandgap crystals. Nature 2001, 414, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Astratov, V.N.; Adawi, A.M.; Fricker, S.; Skolnick, M.S.; Whittaker, D.M.; Pusey, P.N. Interplay of order and disorder in the optical properties of opal photonic crystals. Phys. Rev. B 2002, 66, 165215. [Google Scholar] [CrossRef]
- Von Freymann, G.; Kitaev, V.; Lotschz, B.; Ozin, G. Bottom-up assembly of photonic crystals. Chem. Soc. Rev. 2013, 42, 2528–2554. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; He, L.; Xu, W.; Wang, X.; Yin, Y. Magnetic assembly and field-tuning of ellipsoidal-nanoparticle-based colloidal photonic crystals. Angew. Chem. Int. Ed. 2015, 54, 7077–7081. [Google Scholar] [CrossRef] [PubMed]
- Yethiraj, A.; Thijssen, J.; Wouterse, A.; van Blaaderen, A. Large-area electric-field-induced colloidal single crystals for photonic applications. Adv. Mater. 2004, 16, 596–600. [Google Scholar] [CrossRef]
- Arsenault, A.; Puzzo, D.; Manners, I.; Ozin, G. Photonic-crystal full-colour displays. Nat. Photonics 2007, 1, 468–472. [Google Scholar] [CrossRef]
- Michaelis, B.; Snoswell, D.R.E.; Bell, N.A.W.; Spahn, P.; Hellmann, G.P.; Finlayson, C.E.; Baumberg, J.J. Generating lithographically-defined tunable printed structural color. Adv. Eng. Mater. 2013, 15, 948–953. [Google Scholar] [CrossRef]
- Vowinkel, S.; Schafer, G.; Cherkashinin, G.; Fasel, C.; Roth, F.; Liu, N.; Dietz, C.; Ionescu, E.; Gallei, M. 3d-ordered carbon materials by melt-shear organization for tailor-made hybrid core-shell polymer particle architectures. J. Mater. Chem. C 2016, 4, 3976–3986. [Google Scholar] [CrossRef]
- Schafer, C.G.; Smolin, D.A.; Hellmann, G.P.; Gallei, M. Fully reversible shape transition of soft spheres in elastomeric polymer opal films. Langmuir 2013, 29, 11275–11283. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Redondo, E.; Schmitt, M.; Urbach, Z.; Hui, C.; Sainidou, R.; Rembert, P.; Matyjaszewski, K.; Bockstaller, M.; Fytas, G. A new class of tunable hypersonic phononic crystals based on polymer-tethered colloids. Nat. Commun. 2015, 6, 8309. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Chen, Z.; Zhao, Z.; Wang, H.; Shang, L.; Gua, Z.; Zhao, Y. Bio-inspired self-healing structural color hydrogel. In Proceedings of the National Academy of Science; PNAS: Washington, DC, USA, 2017. [Google Scholar] [CrossRef]
- Finlayson, C.E.; Goddard, C.; Papachristodoulou, E.; Snoswell, D.R.E.; Kontogeorgos, A.; Spahn, P.; Hellmann, G.P.; Hess, O.; Baumberg, J.J. Ordering in stretch-tunable polymeric opal fibers. Opt. Express 2011, 19, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Zhao, Q.; Smoukov, S.; Baumberg, J. Selectively patterning polymer opal films via microimprint lithography. Adv. Opt. Mater. 2014, 2, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Snoswell, D.R.E.; Baumberg, J.J. Stretching the imagination. Textiles 2009, 4, 8–10. [Google Scholar]
- Sussman, J.; Snoswell, D.; Kontogeorgos, A.; Baumberg, J.; Spahn, P. Thermochromic polymer opals. Appl. Phys. Lett. 2009, 95, 173116. [Google Scholar] [CrossRef]
- Braun, P. Materials science colour without colourants. Nature 2011, 472, 423–424. [Google Scholar] [CrossRef] [PubMed]
- Kontogeorgos, A.; Snoswell, D.R.E.; Finlayson, C.E.; Baumberg, J.J.; Spahn, P.; Hellmann, G.P. Inducing symmetry breaking in nanostructures: Anisotropic stretch-tuning photonic crystals. Phys. Rev. Lett. 2010, 105, 233909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Finlayson, C.E.; Snoswell, D.R.E.; Haines, A.I.; Shafer, C.; Spahn, P.; Hellmann, G.P.; Petukhov, A.; Herrmann, L.; Burdet, P.; et al. Large-scale ordering of nanoparticles using viscoelastic shear processing. Nat. Commun. 2016, 7, 11661. [Google Scholar] [CrossRef] [PubMed]
- Ackerson, B.; Pusey, P. Shear-induced order in suspensions of hard-spheres. Phys. Rev. Lett. 1988, 61, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Asher, S.; Weissman, J.; Tikhonov, A.; Coalson, R.; Kesavamoorthy, R. Diffraction in crystalline colloidal-array photonic crystals. Phys. Rev. E 2004, 69, 066619. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D. Order through disorder—Entropy strikes back. Phys. World 1993, 6, 24–25. [Google Scholar] [CrossRef]
- Haw, M.; Poon, W.; Pusey, P. Direct observation of oscillatory-shear-induced order in colloidal suspensions. Phys. Rev. E 1998, 57, 6859–6864. [Google Scholar] [CrossRef]
- Lander, B.; Seifert, U.; Speck, T. Crystallization in a sheared colloidal suspension. J. Chem. Phys. 2013, 138, 224907. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Derks, D.; van Blaaderen, A.; Imhof, A. Melting and crystallization of colloidal hard-sphere suspensions under shear. Proc. Nat. Acad. Sci. USA 2009, 106, 10564–10569. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Finlayson, C.; Schaefer, C.; Spahn, P.; Gallei, M.; Herrmann, L.; Petukhov, A.; Baumberg, J. Nanoassembly of polydisperse photonic crystals based on binary and ternary polymer opal alloys. Adv. Opt. Mater. 2016, 4, 1494–1500. [Google Scholar] [CrossRef]
- Ruhl, T.; Spahn, P.; Hellmann, G.P. Artificial opals prepared by melt compression. Polymer 2003, 44, 7625–7634. [Google Scholar] [CrossRef]
- Spahn, P.; Finlayson, C.E.; Etah, W.M.; Snoswell, D.R.E.; Baumberg, J.J.; Hellmann, G.P. Modification of the refractive-index contrast in polymer opal films. J. Mater. Chem. 2011, 21, 8893–8897. [Google Scholar] [CrossRef]
- Pursiainen, O.; Baumberg, J.; Winkler, H.; Viel, B.; Spahn, P.; Ruhl, T. Nanoparticle-tuned structural color from polymer opals. Opt. Express 2007, 15, 9553–9561. [Google Scholar] [CrossRef] [PubMed]
- Finlayson, C.E.; Spahn, P.; Snoswell, D.R.E.; Yates, G.; Kontogeorgos, A.; Haines, A.I.; Hellmann, G.P.; Baumberg, J.J. 3d bulk ordering in macroscopic solid opaline films by edge-induced rotational shearing. Adv. Mater. 2011, 23, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Mackley, M.; Butler, S.; Baumberg, J.; Snoswell, D.; Finlayson, C.E.; Zhao, Q.B. The rheology and processing of “Edge sheared” Colloidal polymer opals. J. Rheol. 2014, 58, 397–409. [Google Scholar] [CrossRef]
- Senjanovic, I.; Vladimir, N. Physical insight into timoshenko beam theory and its modification with extension. Struct. Eng. Mech. 2013, 48, 519–545. [Google Scholar] [CrossRef]
- Snoswell, D.; Kontogeorgos, A.; Baumberg, J.; Lord, T.; Mackley, M.; Spahn, P.; Hellmann, G. Shear ordering in polymer photonic crystals. Phys. Rev. E 2010, 81, 020401. [Google Scholar] [CrossRef] [PubMed]
- Pursiainen, O.; Baumberg, J.; Winkler, H.; Viel, B.; Spahn, P.; Ruhl, T. Shear-induced organization in flexible polymer opals. Adv. Mater. 2008, 20, 1484–1487. [Google Scholar] [CrossRef]
- Snoswell, D.R.E.; Finlayson, C.E.; Zhao, Q.; Baumberg, J.J. Real-time measurements of crystallization processes in viscoelastic polymeric photonic crystals. Phys. Rev. E 2015, 92, 052315. [Google Scholar] [CrossRef] [PubMed]
- Catherall, A.; Melrose, J.; Ball, R. Shear thickening and order-disorder effects in concentrated colloids at high shear rates. J. Rheol. 2000, 44, 1–25. [Google Scholar] [CrossRef]
- Haines, A.I.; Finlayson, C.E.; Snoswell, D.R.E.; Spahn, P.; Hellmann, G.P.; Baumberg, J.J. Anisotropic resonant scattering from polymer photonic crystals. Adv. Mater. 2012, 24, Op305–Op308. [Google Scholar] [CrossRef] [PubMed]
- Baumberg, J.; Pursiainen, O.; Spahn, P. Resonant optical scattering in nanoparticle-doped polymer photonic crystals. Phys. Rev. B 2009, 80, 201103. [Google Scholar] [CrossRef]
- Finlayson, C.E.; Haines, A.I.; Snoswell, D.R.E.; Kontogeorgos, A.; Vignolini, S.; Baumberg, J.J.; Spahn, P.; Hellmann, G.P. Interplay of index contrast with periodicity in polymer photonic crystals. Appl. Phys. Lett. 2011, 99, 261913. [Google Scholar] [CrossRef]
- Panine, P.; Narayanan, T.; Vermant, J.; Mewis, J. Structure and rheology during shear-induced crystallization of a latex suspension. Phys. Rev. E 2002, 66, 022401. [Google Scholar] [CrossRef] [PubMed]
- Besseling, T.; Hermes, M.; Fortini, A.; Dijkstra, M.; Imhof, A.; van Blaaderen, A. Oscillatory shear-induced 3d crystalline order in colloidal hard-sphere fluids. Soft Matter 2012, 8, 6931–6939. [Google Scholar] [CrossRef]
- Available online: http://www.3ds.com/products-services/simulia/products/abaqus/ (accessed on 21 June 2017).
- Shereda, L.; Larson, R.; Solomon, M. Boundary-driven colloidal crystallization in simple shear flow. Phys. Rev. Lett. 2010, 105, 228302. [Google Scholar] [CrossRef] [PubMed]
- Cates, M.; Wittmer, J.; Bouchaud, J.; Claudin, P. Jamming, force chains, and fragile matter. Phys. Rev. Lett. 1998, 81, 1841–1844. [Google Scholar] [CrossRef]
- Dai, Z.F.; Li, Y.; Duan, G.T.; Jia, L.C.; Cai, W.P. Phase diagram, design of monolayer binary colloidal crystals, and their fabrication based on ethanol-assisted self-assembly at the air/water interface. ACS Nano 2012, 6, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, R.; Mittleman, D.; Rich, C.; Vicki, C. Effect of disorder on the optical properties of colloidal crystals. Phys. Rev. E 2005, 71, 016615. [Google Scholar] [CrossRef] [PubMed]
- Finlayson, C.E.; Baumberg, J.J. Polymer opals as novel photonic materials. Polym. Int. 2013, 62, 1403–1407. [Google Scholar] [CrossRef]
- Ackerson, B.; Clark, N. Sheared colloidal suspensions. Physica A 1983, 118, 221–249. [Google Scholar] [CrossRef]
- Martin, S.; Bryant, G.; van Megen, W. Observation of a smecticlike crystalline structure in polydisperse colloids. Phys. Rev. Lett. 2003, 90, 255702. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Larsen, R.; Weitz, D. Uniform nonspherical colloidal particles with tunable shapes. Adv. Mater. 2007, 19, 2005–2009. [Google Scholar] [CrossRef]
- Viel, B.; Ruhl, T.; Hellmann, G. Reversible deformation of opal elastomers. Chem. Mater. 2007, 19, 5673–5679. [Google Scholar] [CrossRef]
- Schafer, C.G.; Gallei, M.; Zahn, J.T.; Engelhardt, J.; Hellmann, G.P.; Rehahn, M. Reversible light-, thermo-, and mechano-responsive elastomeric polymer opal films. Chem. Mater. 2013, 25, 2309–2318. [Google Scholar] [CrossRef]
- Zoorob, M.; Charlton, M.; Parker, G.; Baumberg, J.; Netti, M. Complete photonic bandgaps in 12-fold symmetric quasicrystals. Nature 2000, 404, 740–743. [Google Scholar] [PubMed]
- Hynninen, A.P.; Thijssen, J.H.J.; Vermolen, E.C.M.; Dijkstra, M.; Van Blaaderen, A. Self-assembly route for photonic crystals with a bandgap in the visible region. Nat. Mater. 2007, 6, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Finlayson, C.E.; Cattaneo, F.; Perney, N.M.B.; Baumberg, J.J.; Netti, M.C.; Zoorob, M.E.; Charlton, M.D.B.; Parker, G.J. Slow light and chromatic temporal dispersion in photonic crystal waveguides using femtosecond time of flight. Phys. Rev. E 2006, 73, 016619. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finlayson, C.E.; Baumberg, J.J. Generating Bulk-Scale Ordered Optical Materials Using Shear-Assembly in Viscoelastic Media. Materials 2017, 10, 688. https://doi.org/10.3390/ma10070688
Finlayson CE, Baumberg JJ. Generating Bulk-Scale Ordered Optical Materials Using Shear-Assembly in Viscoelastic Media. Materials. 2017; 10(7):688. https://doi.org/10.3390/ma10070688
Chicago/Turabian StyleFinlayson, Chris E., and Jeremy J. Baumberg. 2017. "Generating Bulk-Scale Ordered Optical Materials Using Shear-Assembly in Viscoelastic Media" Materials 10, no. 7: 688. https://doi.org/10.3390/ma10070688
APA StyleFinlayson, C. E., & Baumberg, J. J. (2017). Generating Bulk-Scale Ordered Optical Materials Using Shear-Assembly in Viscoelastic Media. Materials, 10(7), 688. https://doi.org/10.3390/ma10070688