Synthesis of Nanoscale CaO-Al2O3-SiO2-H2O and Na2O-Al2O3-SiO2-H2O Using the Hydrothermal Method and Their Characterization

Abstract
:1. Introduction
2. Experimental Procedures
2.1. Materials
2.2. Synthesis of C-A-S-H and N-A-S-H
2.3. Characterization
3. Results and Discussion
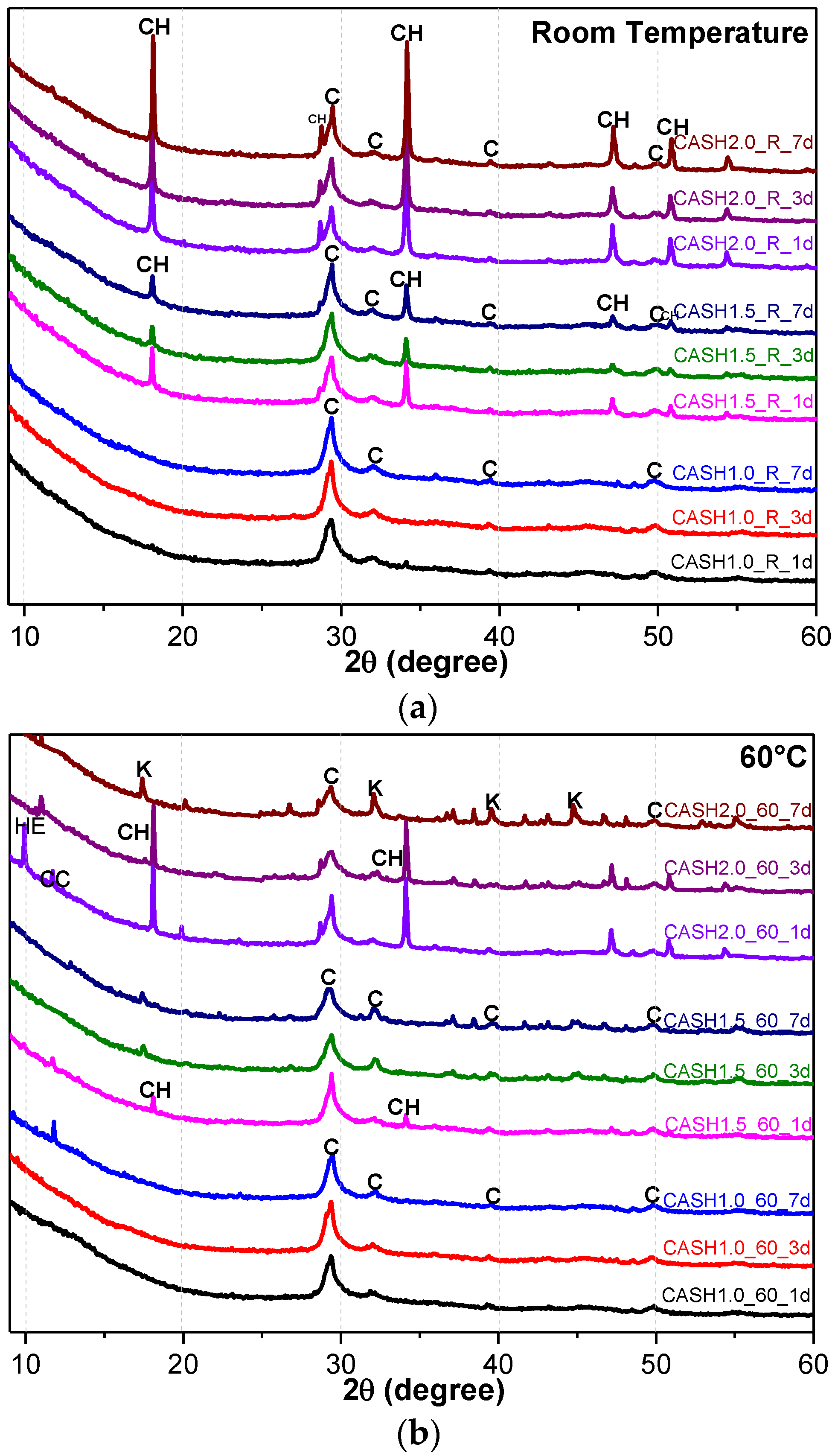
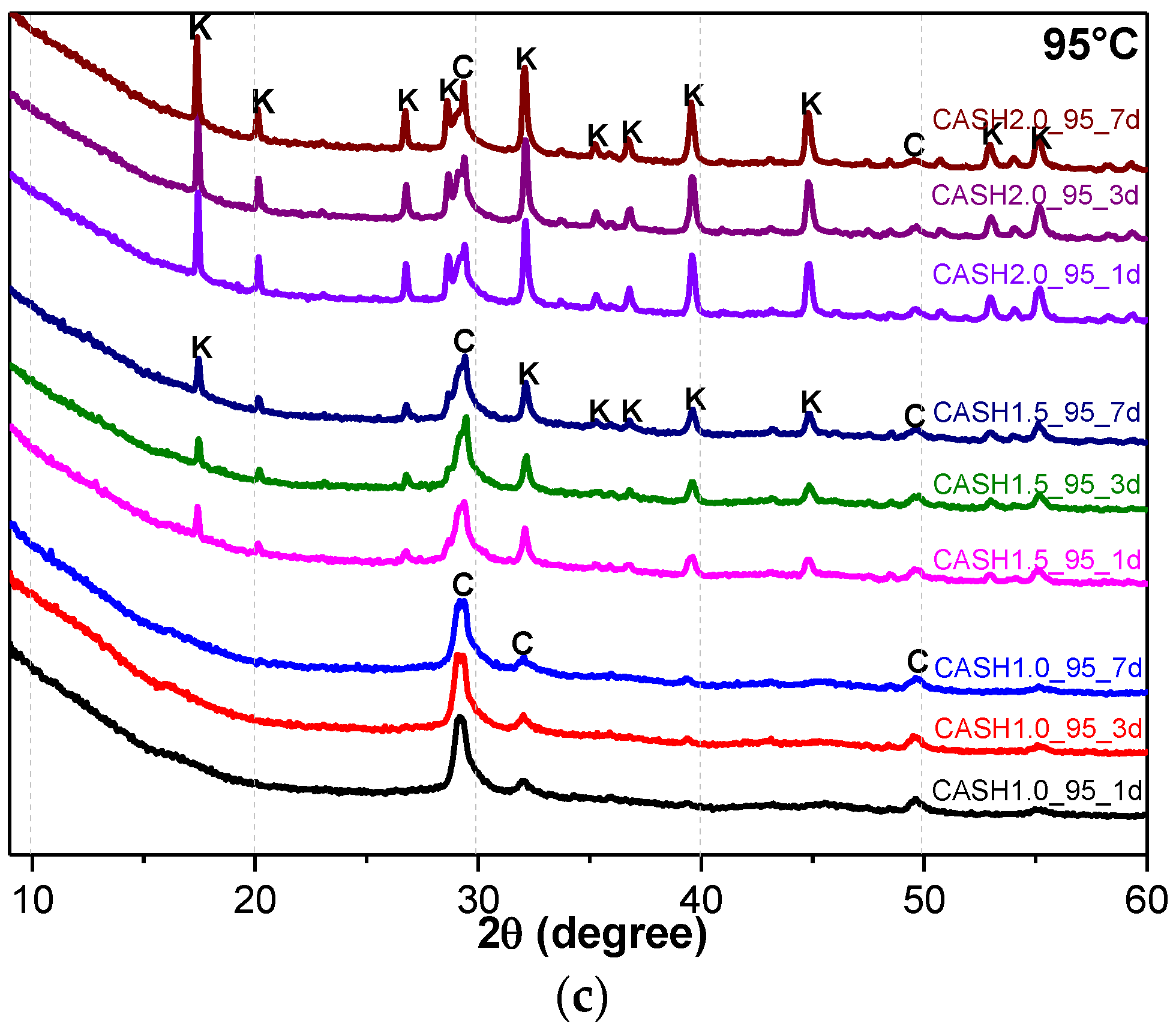
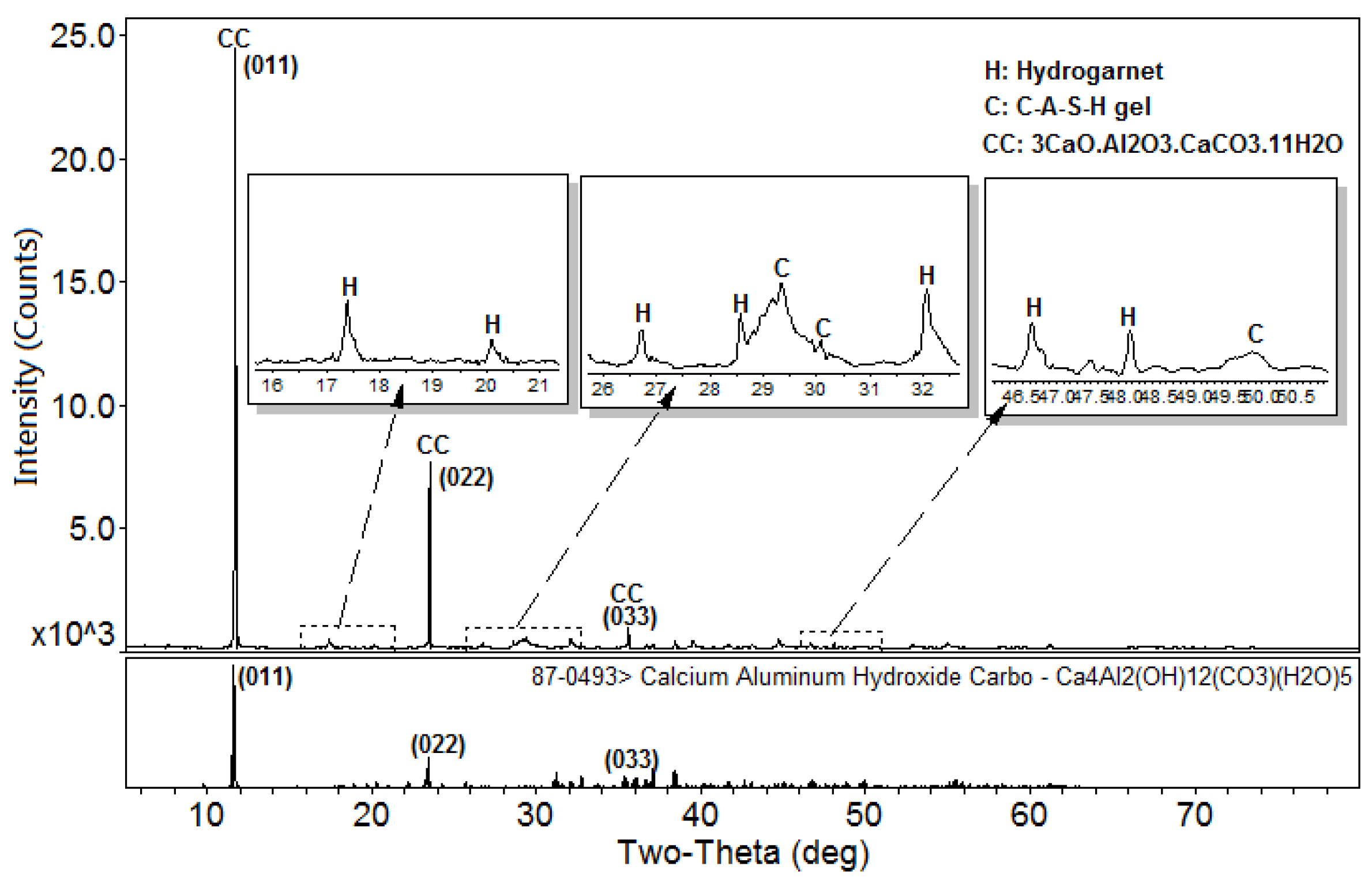
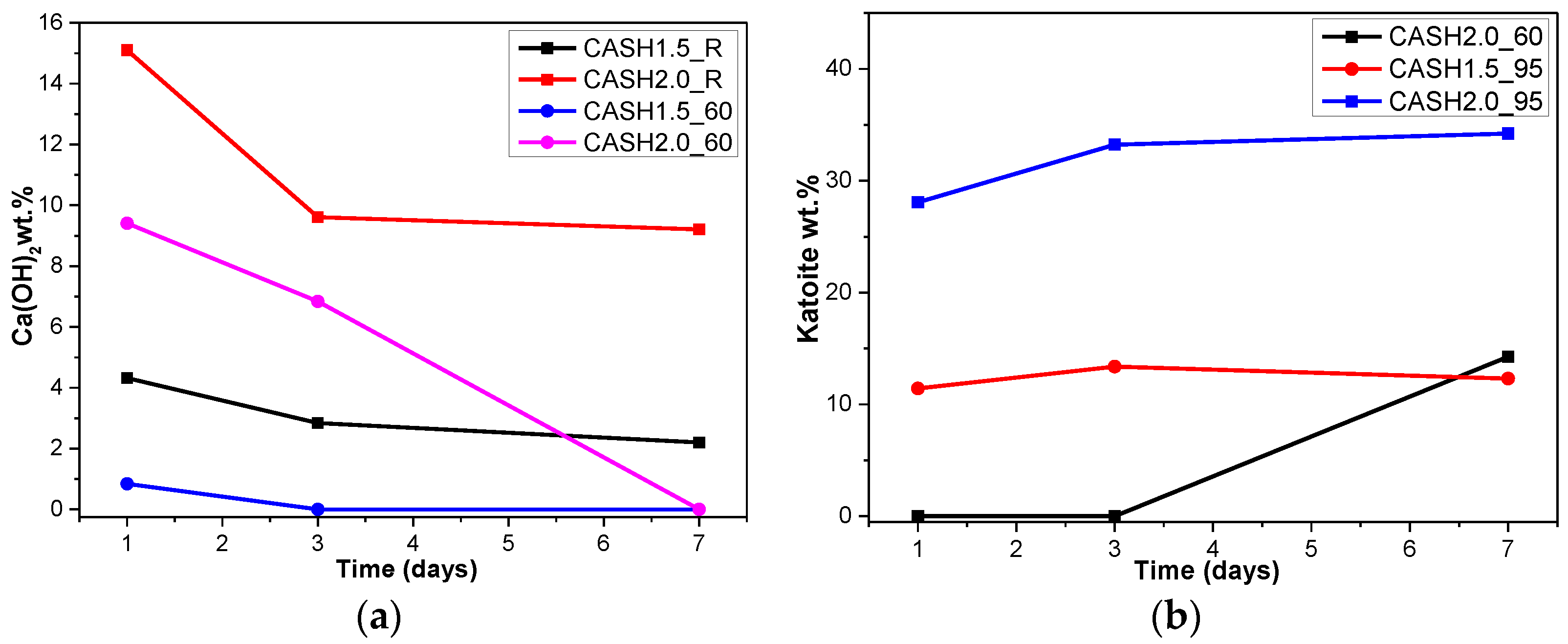
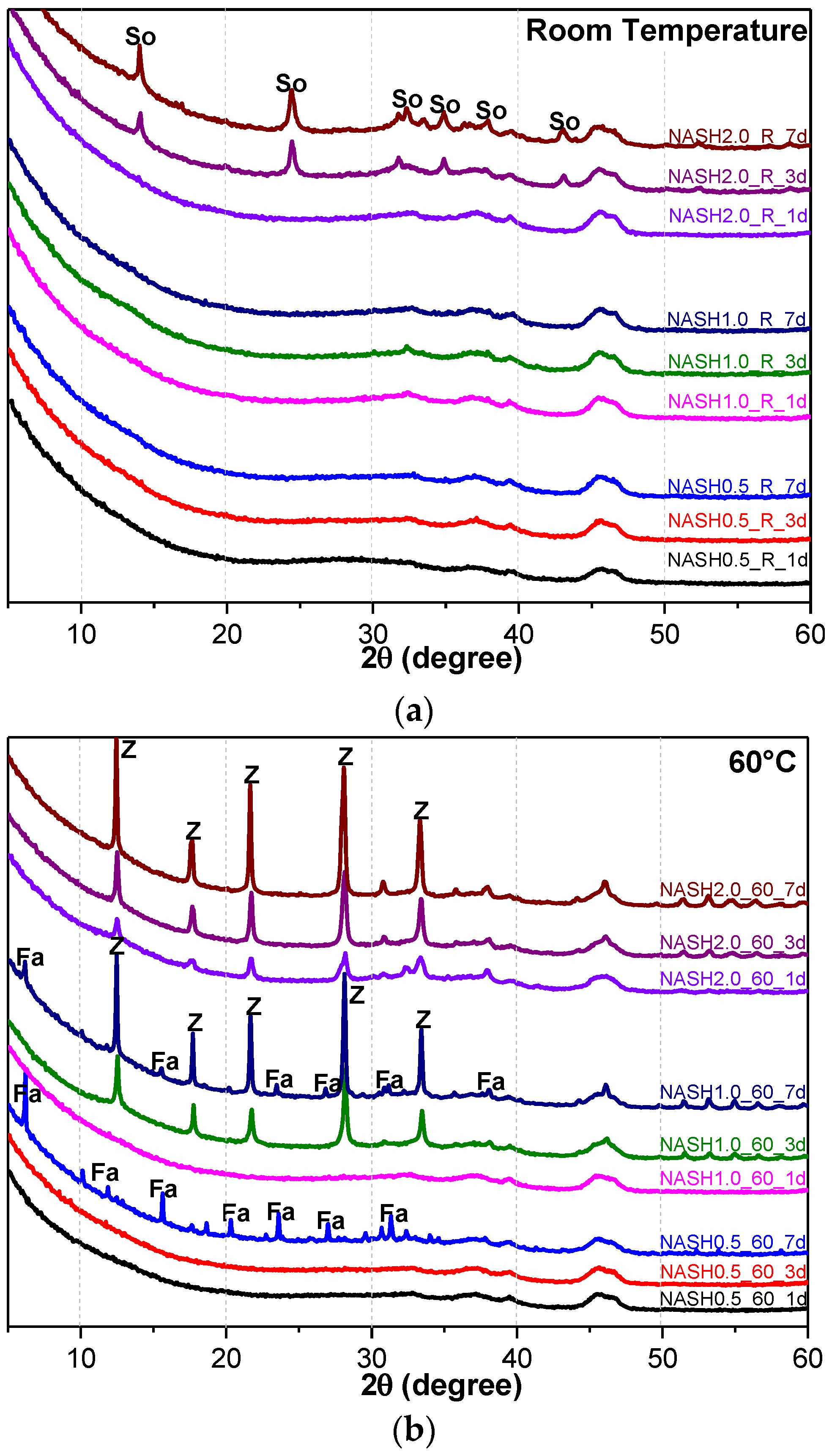
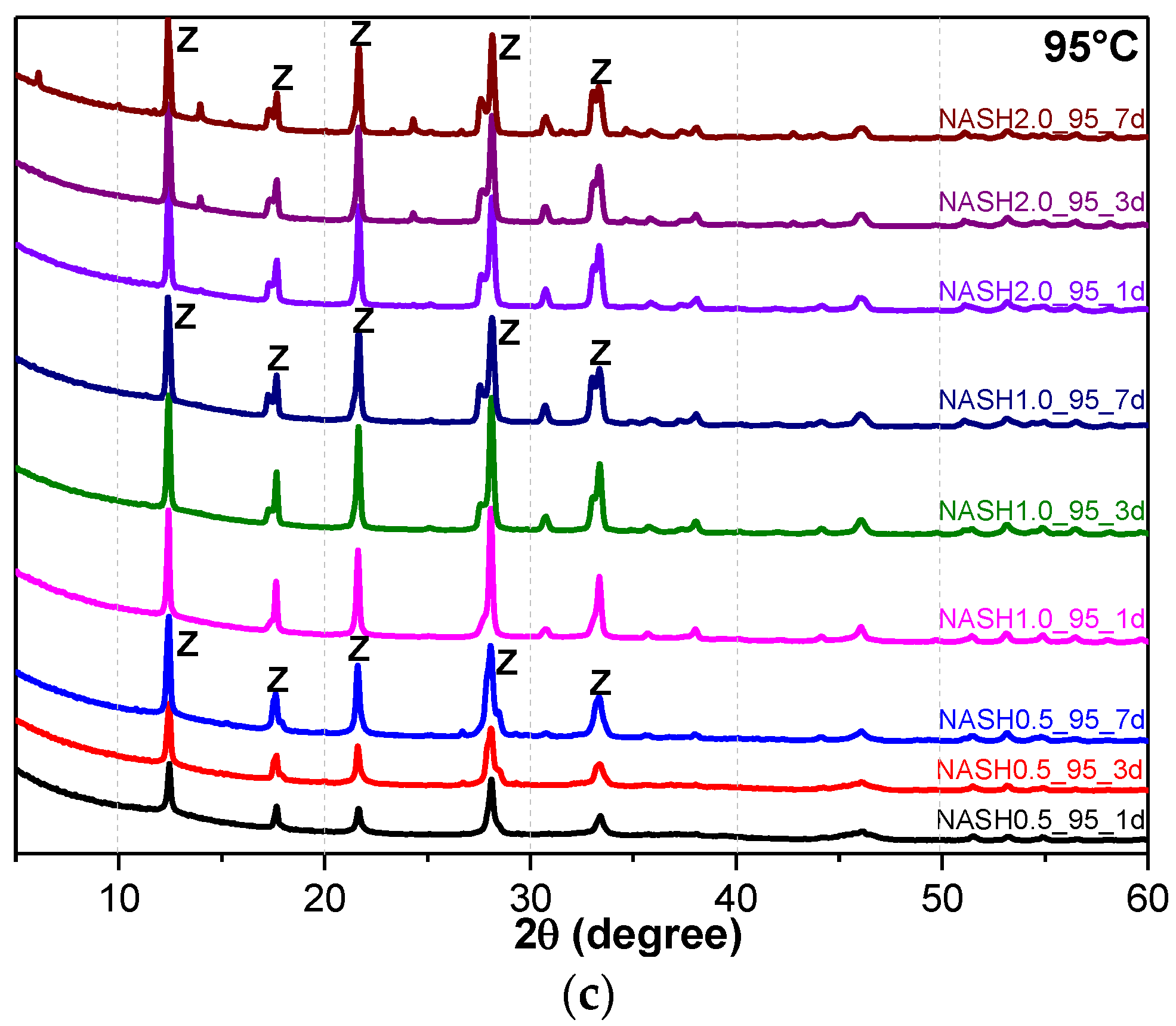
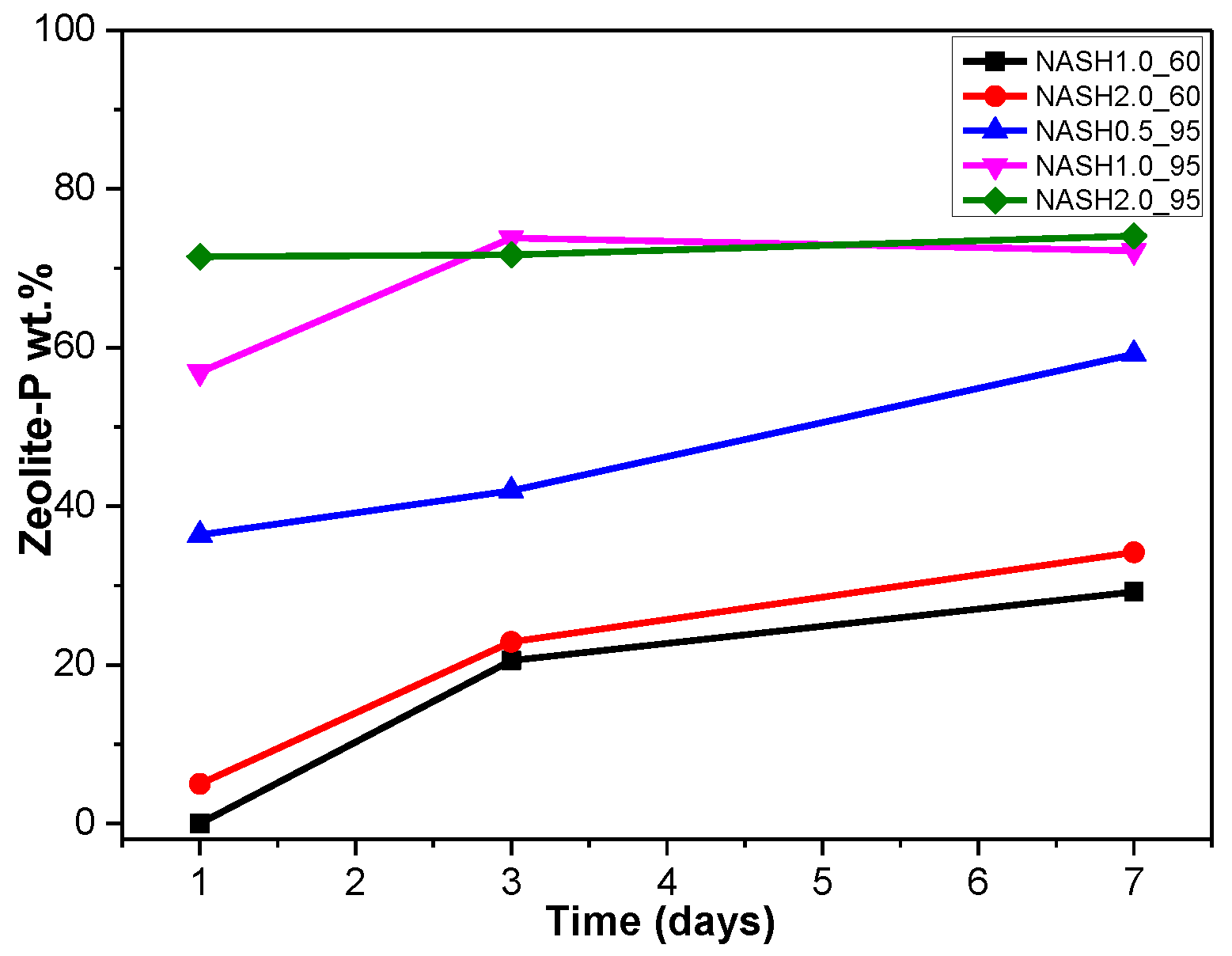
3.1. XRD and Reference Intensity Ratio (RIR) Analysis
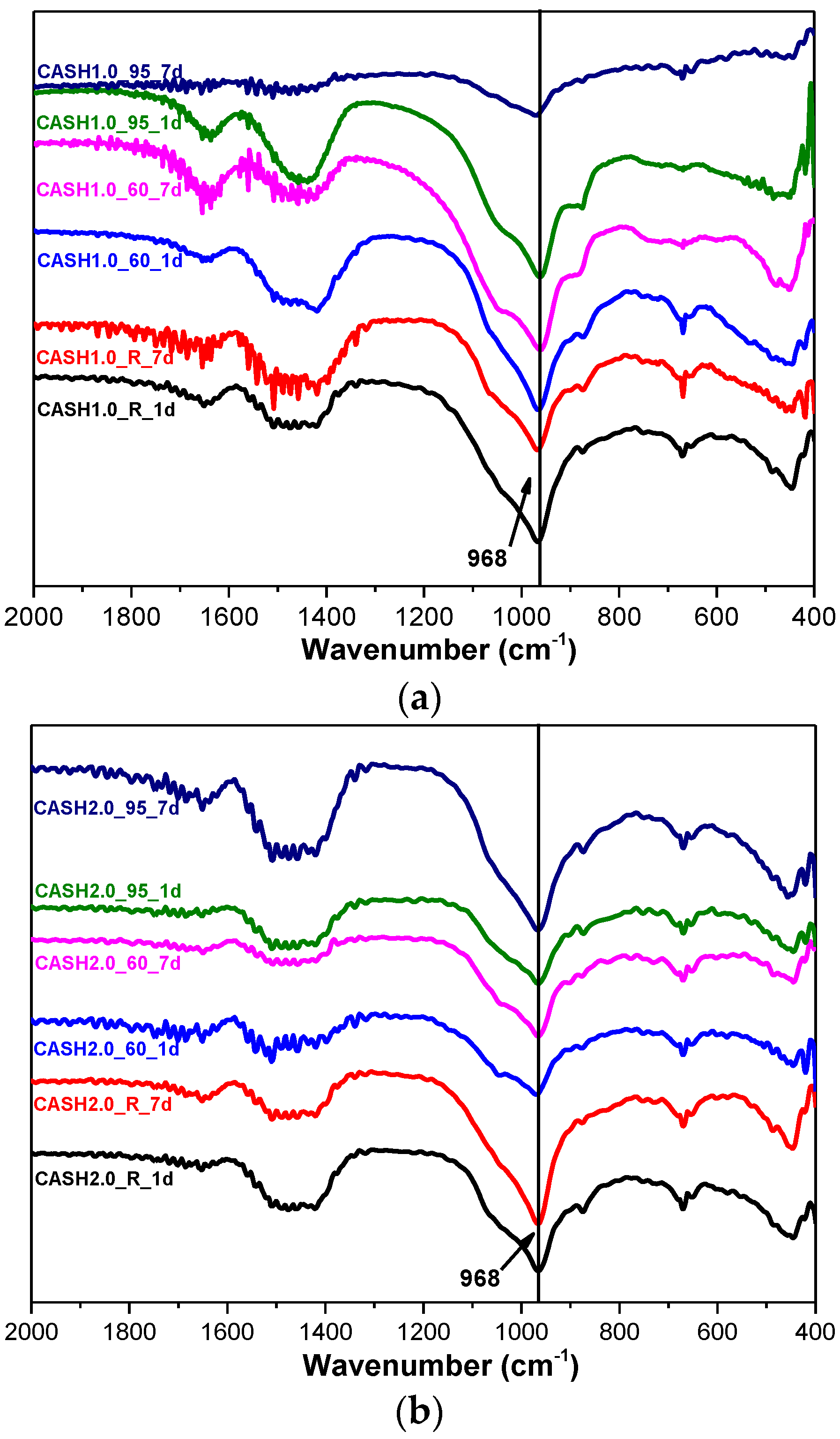
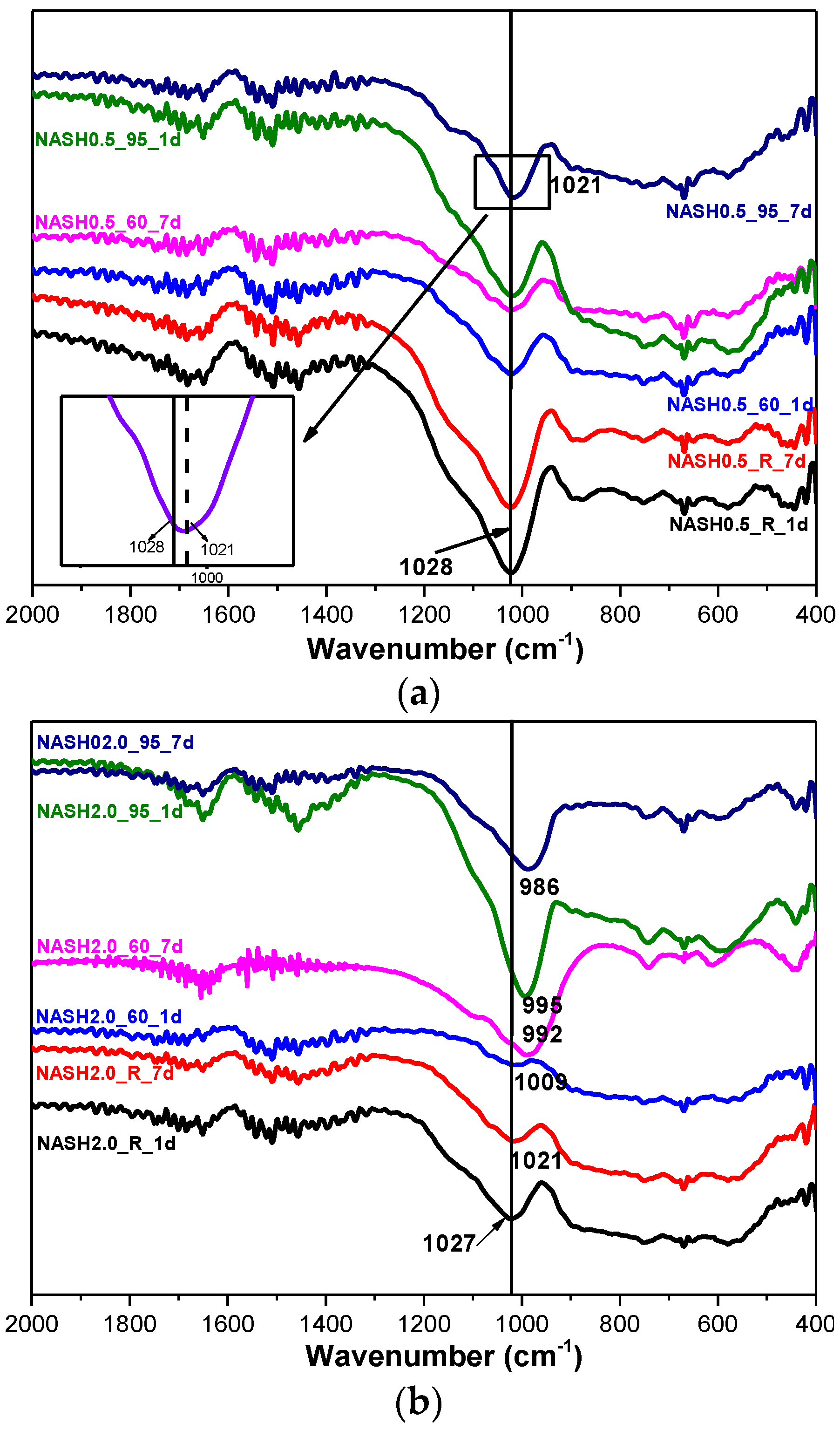
3.2. Fourier Transform Infrared Spectroscopy
3.3. Scanning Electron Microscopy
3.4. Transmission Electron Microscopy
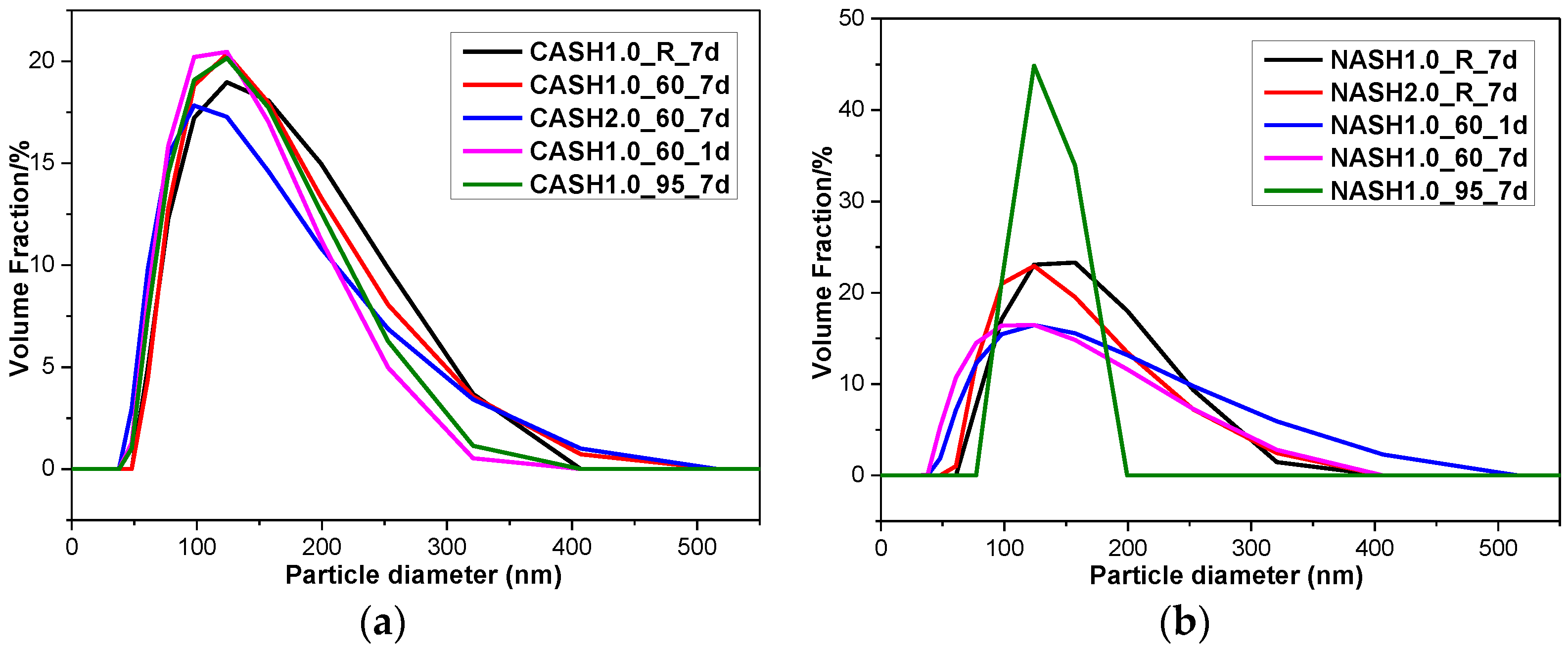
3.5. Particle Size Distribution
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Juenger, M.C.G.; Winnefeld, F.; Provis, J.L.; Ideker, J.H. Advances in alternative cementitious binders. Cem. Concr. Res. 2011, 41, 1232–1243. [Google Scholar] [CrossRef]
- Myers, R.J.; Bernal, S.A.; San, N.R.; Provis, J.L. Generalized structural description of calcium-sodium aluminosilicate hydrate gels: The cross-linked substituted tobermorite model. Langmuir 2013, 29, 5294–5306. [Google Scholar] [CrossRef] [PubMed]
- Puertas, F.; Palacios, M.; Manzano, H.; Dolado, J.S.; Rico, A.; Rodríguez, J. A model for the C-A-S-H gel formed in alkali-activated slag cements. J. Eur. Ceram. Soc. 2011, 31, 2043–2056. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Puertas, F.; Sobrados, I.; Sanz, J. Structure of calcium silicate hydrates formed in alkaline-activated slag: Influence of the type of alkaline activator. J. Am. Ceram. Soc. 2003, 86, 1389–1394. [Google Scholar] [CrossRef]
- Provis, J.L.; Bernal, S.A. Geopolymers and related alkali-activated materials. Mater. Res. 2014, 44, 299–327. [Google Scholar] [CrossRef]
- Sun, G.K.; Young, J.F.; Kirkpatrick, R.J. The role of Al in C-S-H: NMR, XRD, and compositional results for precipitated samples. Cem. Concr. Res. 2006, 36, 18–29. [Google Scholar] [CrossRef]
- Lecomte, I.; Liegeois, M.; Rulmont, A.; Cloots, R.; Maseri, F. Synthesis and characterization of new inorganic polymeric composites based on kaolin or white clay and on ground-granulated blast furnace slag. J. Mater. Res. 2003, 18, 2571–2579. [Google Scholar] [CrossRef]
- Deventer, J.S.J.V.; Provis, J.L.; Duxson, P. Technical and commercial progress in the adoption of geopolymer cement. Miner. Eng. 2012, 29, 89–104. [Google Scholar] [CrossRef]
- Walkley, B.; Nicolas, R.S.; Sani, M.A.; Rees, G.J.; Hanna, J.V.; Deventer, J.S.J.V.; Provis, J.L. Phase evolution of C-(N)-A-S-H/N-A-S-H gel blends investigated via alkali-activation of synthetic calcium aluminosilicate precursors. Cem. Concr. Res. 2016, 89, 120–135. [Google Scholar] [CrossRef]
- Walkley, B.; San, N.R.; Sani, M.A.; Gehman, J.D.; van Deventer, J.S.; Provis, J.L. Phase evolution of Na2O-Al2O3-SiO2-H2O gels in synthetic aluminosilicate binders. Dalton Trans. 2016, 45, 5521–5535. [Google Scholar] [CrossRef] [PubMed]
- L’Hôpital, E.; Lothenbach, B.; Le Saout, G.; Kulik, D.; Scrivener, K. Incorporation of aluminium in calcium-silicate-hydrates. Cem. Concr. Res. 2015, 75, 91–103. [Google Scholar] [CrossRef]
- L’Hôpital, E.; Lothenbach, B.; Kulik, D.A.; Scrivener, K. Influence of calcium to silica ratio on aluminium uptake in calcium silicate hydrate. Cem. Concr. Res. 2016, 85, 111–121. [Google Scholar] [CrossRef]
- García-Lodeiro, I.; Fernández-Jiménez, A.; Palomo, A.; Macphee, D.E. Effect of calcium additions on N-A-S-H cementitious gels. J. Am. Ceram. Soc. 2010, 93, 1934–1940. [Google Scholar] [CrossRef]
- García-Lodeiro, I.; Fernández-Jiménez, A.; Blanco, M.T.; Palomo, A. FTIR study of the sol-gel synthesis of cementitious gels: C-S-H and N-A-S-H. J. Sol-Gel Sci. Technol. 2008, 45, 63–72. [Google Scholar] [CrossRef]
- Cui, X.; Liu, L.; Zheng, G.; Wang, R.; Lu, J. Characterization of chemosynthetic Al2O3-2SiO2 geopolymers. J. Non-Cryst. Solids 2010, 356, 72–76. [Google Scholar] [CrossRef]
- Julie, R.; Fabien, F.; Celine, C.D.C.; Annie, M.; Thierry, D.; Christophe, J.D. Incorporation of aluminum into C-S-H structures: from synthesis to nanostructural characterization. J. Am. Ceram. Soc. 2008, 91, 2337–2342. [Google Scholar]
- Hartmann, A.; Buhl, J.C.; Breugel, K.V. Structure and phase investigations on crystallization of 11 Å tobermorite in lime sand pellets. Cem. Concr. Res. 2007, 37, 21–31. [Google Scholar] [CrossRef]
- Matsui, K.; Kikuma, J.; Tsunashima, M.; Ishikawa, T.; Matsuno, S.Y.; Ogawa, A.; Sato, M. In situ time-resolved X-ray diffraction of tobermorite formation in autoclaved aerated concrete: Influence of silica source reactivity and Al addition. Cem. Concr. Res. 2011, 41, 510–519. [Google Scholar] [CrossRef]
- Shi, C.; Krivenko, P.; Roy, D. Alkali-Activated Cements and Concretes; Taylor & Francis: Abingdon, UK, 2005. [Google Scholar]
- Myers, R.J. Thermodynamic Modelling of CaO-Al2O3-SiO2-H2O-Based Cements. Ph.D. Thesis, The University of Sheffield, Sheffield, UK, June 2015. [Google Scholar]
- Renaudin, G.; Russias, J.; Leroux, F.; Frizon, F.; Cau-dit-Coumes, C. Structural characterization of C-S-H and C-A-S-H samples-Part I: Long-range order investigated by Rietveld analyses. J. Solid State Chem. 2009, 182, 3312–3319. [Google Scholar] [CrossRef]
- Baltakys, K.; Eisinas, A.; Doneliene, J.; Monstvilaite, D.; Bankauskaite, A.; Urbutis, A. The hydrothermal synthesis of calcium aluminium silicate hydrates and its application on Portland cement hydration. Rev. Rom. Mater. 2015, 45, 218–225. [Google Scholar]
- Walkley, B.; Provis, J.L.; San Nicolas, R.; Sani, M.A.; van Deventer, J.S.J. Stoichiometrically controlled C-(A)-S-H/N-A-S-H gel blends via alkali-activation of synthetic precursors. Adv. Appl. Ceram. 2015, 114, 372–377. [Google Scholar] [CrossRef]
- Bernal, S.A.; Provis, J.L.; Walkley, B.; San Nicolas, R.; Gehman, J.D.; Brice, D.G.; Kilcullen, A.R.; Duxson, P.; van Deventer, J.S.J. Gel nanostructure in alkali-activated binders based on slag and fly ash, and effects of accelerated carbonation. Cem. Concr. Res. 2013, 53, 127–144. [Google Scholar] [CrossRef]
- Bernal, S.A.; San Nicolas, R.; Provis, J.L.; Mejía De Gutiérrez, R.; van Deventer, J.S.J. Natural carbonation of aged alkali-activated slag concretes. Mater. Struct. 2014, 47, 693–707. [Google Scholar] [CrossRef]
- Bernal, S.A.; Provis, J.L.; Myers, R.J.; San Nicolas, R.; van Deventer, J.S.J. Role of carbonates in the chemical evolution of sodium carbonate-activated slag binders. Mater. Struct. 2015, 48, 517–529. [Google Scholar] [CrossRef]
- Chung, F.H. Quantitative interpretation of X-ray diffraction patterns of mixtures. I. Matrix-flushing method for quantitative multicomponent analysis. J. Appl. Cryst. 1974, 7, 519–525. [Google Scholar] [CrossRef]
- Rojas, M.F.A.; Cabrera, J. The effect of temperature on the hydration rate and stability of the hydration phases of metakaolin-lime-water systems. Cem. Concr. Res. 2002, 32, 133–138. [Google Scholar] [CrossRef]
- Oh, J.E.; Moon, J.; Mancio, M.; Clark, S.M.; Monteiro, P.J.M. Bulk modulus of basic sodalite, Na8[AlSiO4]6(OH)2·2H2O, a possible zeolitic precursor in coal-fly-ash-based geopolymers. Cem. Concr. Res. 2011, 41, 107–112. [Google Scholar] [CrossRef]
- Criado, M.; Fernández-Jiménez, A.; de la Torre, A.G.; Aranda, M.A.G.; Palomo, A. An XRD study of the effect of the SiO2/Na2O ratio on the alkali activation of fly ash. Cem. Concr. Res. 2007, 37, 671–679. [Google Scholar] [CrossRef]
- Istuque, D.B.; Reig, L.; Moraes, J.C.B.; Akasaki, J.L.; Borrachero, M.V.; Soriano, L.; Payá, J.; Malmonge, J.A.; Tashima, M.M. Behaviour of metakaolin-based geopolymers incorporating sewage sludge ash (SSA). Mater. Lett. 2016, 180, 192–195. [Google Scholar] [CrossRef]
- Garcia-Lodeiro, I.; Palomo, A.; Macphee, D.E. Compatibility studies between N-A-S-H and C-A-S-H gels. Study in the ternary diagram Na2O-CaO-Al2O3-SiO2-H2O. Cem. Concr. Res. 2011, 41, 923–931. [Google Scholar] [CrossRef]
- Andini, S.; Cioffi, R.; Colangelo, F.; Grieco, T.; Montagnaro, F.; Santoro, L. Coal fly ash as raw material for the manufacture of geopolymer-based products. Waste Manag. 2008, 28, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Skvára, F.; Kopecký, L.; Smilauer, V.; Bittnar, Z. Material and structural characterization of alkali activated low-calcium brown coal fly ash. J. Hazard Mater. 2009, 168, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.K.W.; Deventer, J.S.J.V. Use of infrared spectroscopy to study geopolymerization of heterogeneous amorphous aluminosilicates. Langmuir 2003, 19, 8726–8734. [Google Scholar] [CrossRef]
- Fujita, S.; Sakamoto, A.; Tomozawa, M. Fictive temperature measurement of alumino-silicate glasses using IR spectroscopy. J. Non-Cryst. Solids 2003, 330, 252–258. [Google Scholar] [CrossRef]
- Garcia-Lodeiro, I.; Fernández-Jimenez, A.; Pena, P.; Palomo, A. Alkaline activation of synthetic aluminosilicate glass. Ceram. Int. 2014, 40, 5547–5558. [Google Scholar] [CrossRef]
- Passaglia, E.; RinaldiI, R. Katoite, a new member of the Ca3Al2(SiO4)3-Ca3Al2(SiO4)3(OH)12 series and a new nomenclature for the hydrogrossular group of minerals. Bull. Mineral. 1984, 107, 605–618. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CaO/SiO2 | Na2O/SiO2 | SiO2/Al2O3 | Water/Solid Ratio |
|---|---|---|---|---|
| CASH1.0 | 1.0 | - | 2.0 | 8.0 |
| CASH1.5 | 1.5 | - | 2.0 | 8.0 |
| CASH2.0 | 2.0 | - | 2.0 | 8.0 |
| NASH0.5 | - | 0.5 | 2.0 | 5.0 |
| NASH1.0 | - | 1.0 | 2.0 | 5.0 |
| NASH2.0 | - | 2.0 | 2.0 | 5.0 |
| Sample | Result (wt.%) | Sample | Result (wt.%) | Sample | Result (wt.%) |
|---|---|---|---|---|---|
| CASH1.0_R_1d | - | CASH1.0_60_1d | - | CASH1.0_95_1d | - |
| CASH1.0_R_3d | - | CASH1.0_60_3d | - | CASH1.0_95_3d | - |
| CASH1.0_R_7d | - | CASH1.0_60_7d | - | CASH1.0_95_7d | - |
| CASH1.5_R_1d | Ca(OH)2: 4.32 | CASH1.5_60_1d | Ca(OH)2: 0.84 | CASH1.5_95_1d | Katoite: 11.42 |
| CASH1.5_R_3d | Ca(OH)2: 2.84 | CASH1.5_60_3d | - | CASH1.5_95_3d | Katoite: 13.38 |
| CASH1.5_R_7d | Ca(OH)2: 2.20 | CASH1.5_60_7d | - | CASH1.5_95_7d | Katoite: 12.31 |
| CASH2.0_R_1d | Ca(OH)2: 15.10 | CASH2.0_60_1d | Ca(OH)2: 9.41 | CASH2.0_95_1d | Katoite: 28.08 |
| CASH2.0_R_3d | Ca(OH)2: 9.61 | CASH2.0_60_3d | Ca(OH)2: 6.84 | CASH2.0_95_3d | Katoite: 33.23 |
| CASH2.0_R_7d | Ca(OH)2: 9.21 | CASH2.0_60_7d | Katoite: 14.26 | CASH2.0_95_7d | Katoite: 34.22 |
| Sample | Result (wt.%) | Sample | Result (wt.%) | Sample | Result (wt.%) |
|---|---|---|---|---|---|
| NASH0.5_R_1d | - | NASH0.5_60_1d | - | NASH0.5_95_1d | Zeolite-P: 36.40 |
| NASH0.5_R_3d | - | NASH0.5_60_3d | - | NASH0.5_95_3d | Zeolite-P: 41.96 |
| NASH0.5_R_7d | - | NASH0.5_60_7d | Faujasite: 11.62 | NASH0.5_95_7d | Zeolite-P: 59.17 |
| NASH1.0_R_1d | - | NASH1.0_60_1d | - | NASH1.0_95_1d | Zeolite-P: 56.91 |
| NASH1.0_R_3d | - | NASH1.0_60_3d | Zeolite-P: 20.55 | NASH1.0_95_3d | Zeolite-P: 73.82 |
| NASH1.0_R_7d | - | NASH1.0_60_7d | Faujasite: 9.81/Zeolite-P: 29.19 | NASH1.0_95_7d | Zeolite-P: 72.22 |
| NASH2.0_R_1d | - | NASH2.0_60_1d | Zeolite-P: 5.00 | NASH2.0_95_1d | Zeolite-P: 71.45 |
| NASH2.0_R_3d | Sodalite: 5.11 | NASH2.0_60_3d | Zeolite-P: 22.90 | NASH2.0_95_3d | Zeolite-P: 71.68 |
| NASH2.0_R_7d | Sodalite: 9.37 | NASH2.0_60_7d | Zeolite-P: 34.19 | NASH2.0_95_7d | Zeolite-P: 74.08 |
| Sample | Average Particle Diameter (nm) | Sample | Average Particle Diameter (nm) |
|---|---|---|---|
| CASH1.0_R_7d | 295.7 | NASH1.0_R_7d | 304.5 |
| CASH1.0_60_7d | 291.5 | NASH2.0_R_7d | 285.7 |
| CASH2.0_60_7d | 270.2 | NASH1.0_60_1d | 308.3 |
| CASH1.0_60_1d | 253.1 | NASH1.0_60_7d | 261.3 |
| CASH1.0_95_7d | 264.7 | NASH1.0_95_7d | 259.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Li, D.; Fang, Y. Synthesis of Nanoscale CaO-Al2O3-SiO2-H2O and Na2O-Al2O3-SiO2-H2O Using the Hydrothermal Method and Their Characterization. Materials 2017, 10, 695. https://doi.org/10.3390/ma10070695
Yang J, Li D, Fang Y. Synthesis of Nanoscale CaO-Al2O3-SiO2-H2O and Na2O-Al2O3-SiO2-H2O Using the Hydrothermal Method and Their Characterization. Materials. 2017; 10(7):695. https://doi.org/10.3390/ma10070695
Chicago/Turabian StyleYang, Jingbin, Dongxu Li, and Yuan Fang. 2017. "Synthesis of Nanoscale CaO-Al2O3-SiO2-H2O and Na2O-Al2O3-SiO2-H2O Using the Hydrothermal Method and Their Characterization" Materials 10, no. 7: 695. https://doi.org/10.3390/ma10070695
APA StyleYang, J., Li, D., & Fang, Y. (2017). Synthesis of Nanoscale CaO-Al2O3-SiO2-H2O and Na2O-Al2O3-SiO2-H2O Using the Hydrothermal Method and Their Characterization. Materials, 10(7), 695. https://doi.org/10.3390/ma10070695