Study of Superbase-Based Deep Eutectic Solvents as the Catalyst in the Chemical Fixation of CO2 into Cyclic Carbonates under Mild Conditions

 ,
, 
Abstract
:1. Introduction
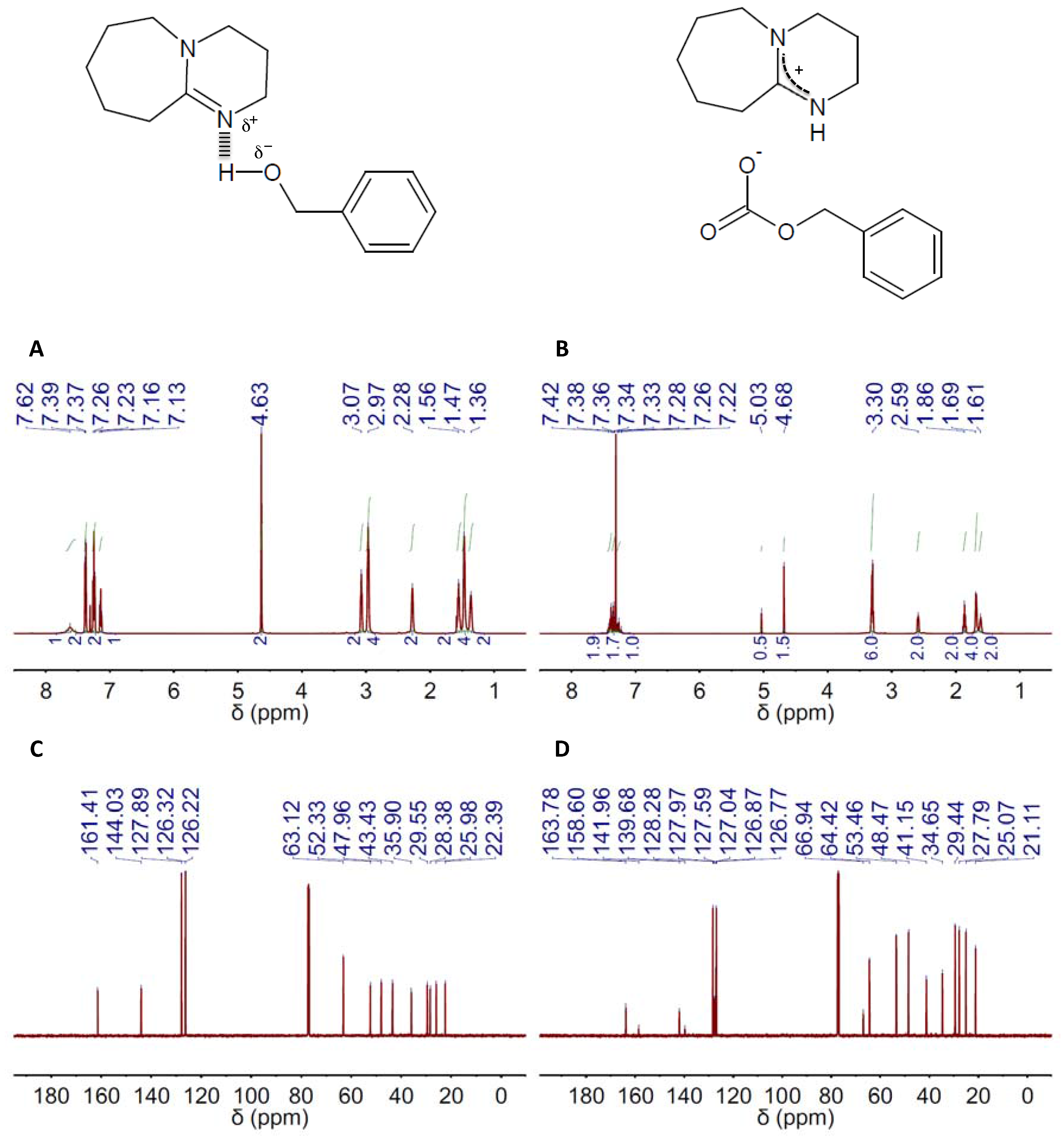
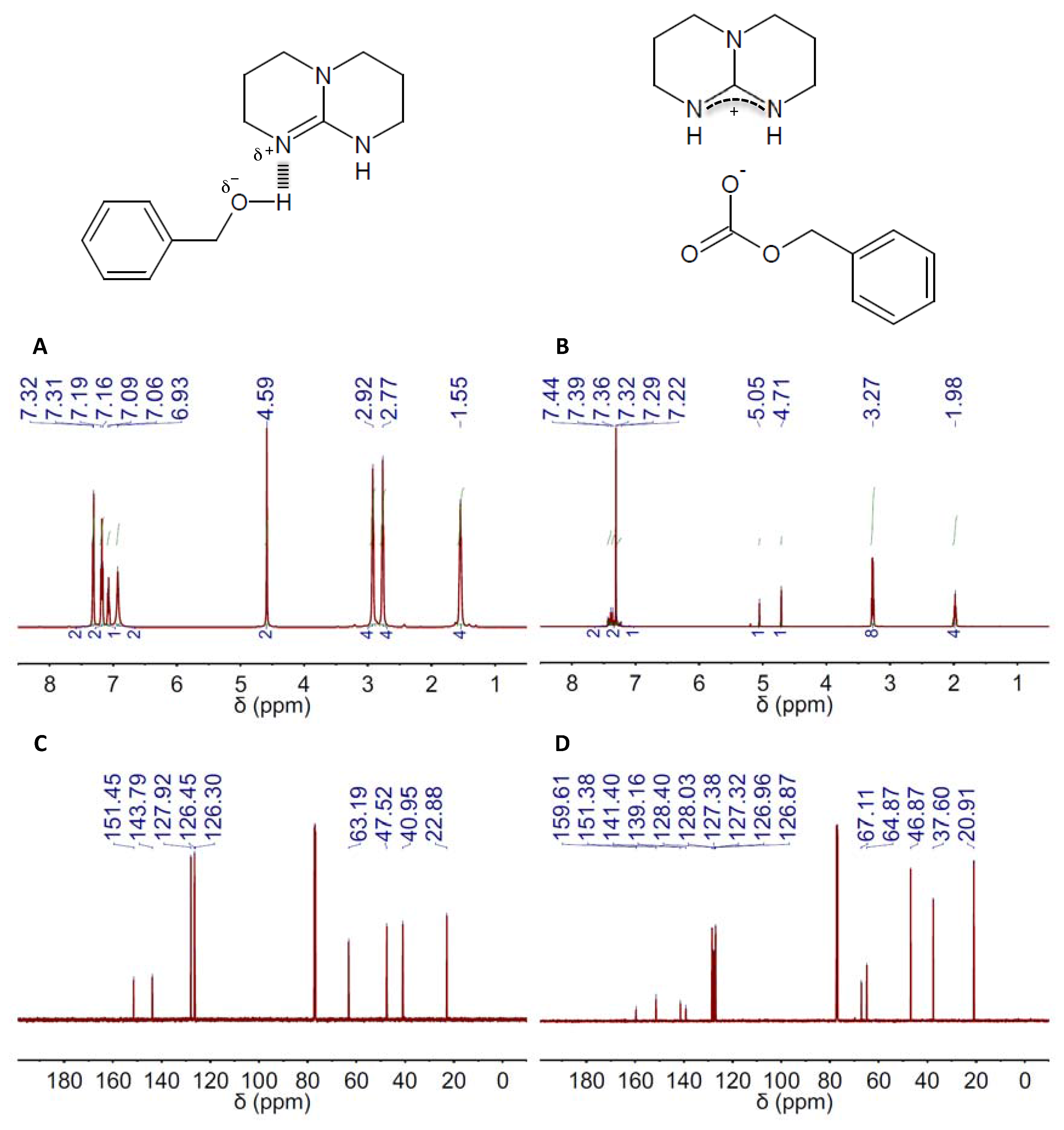
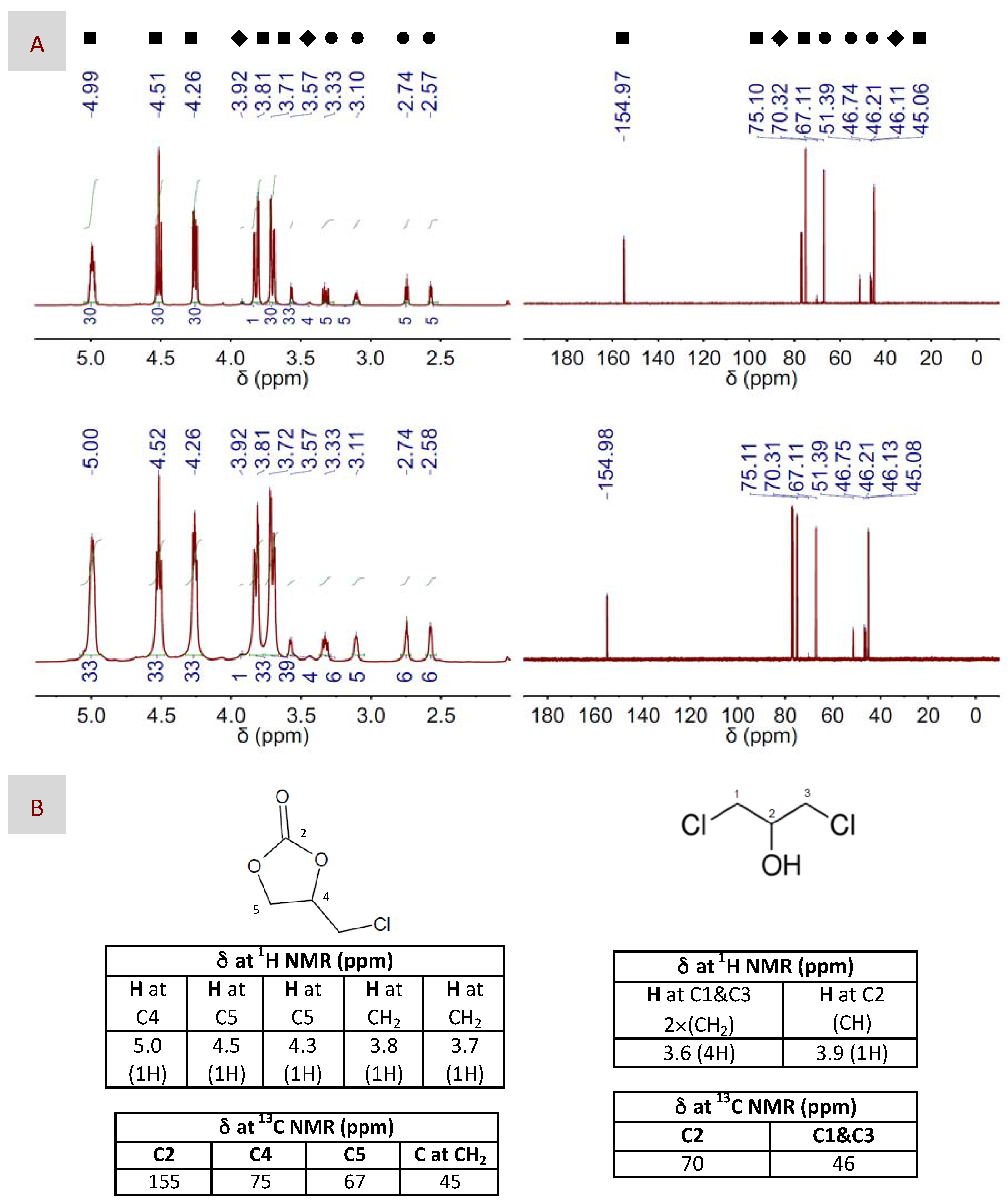
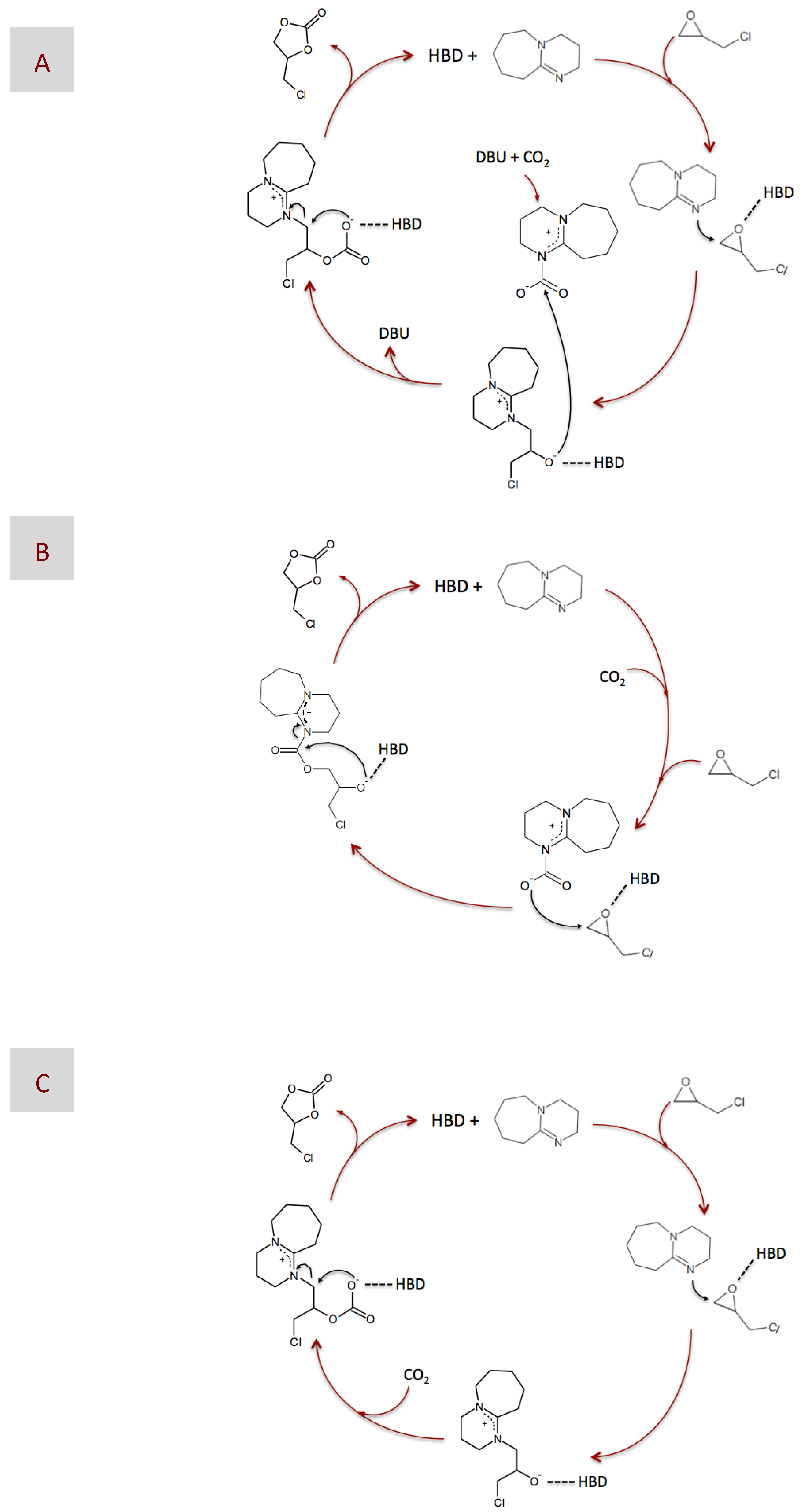
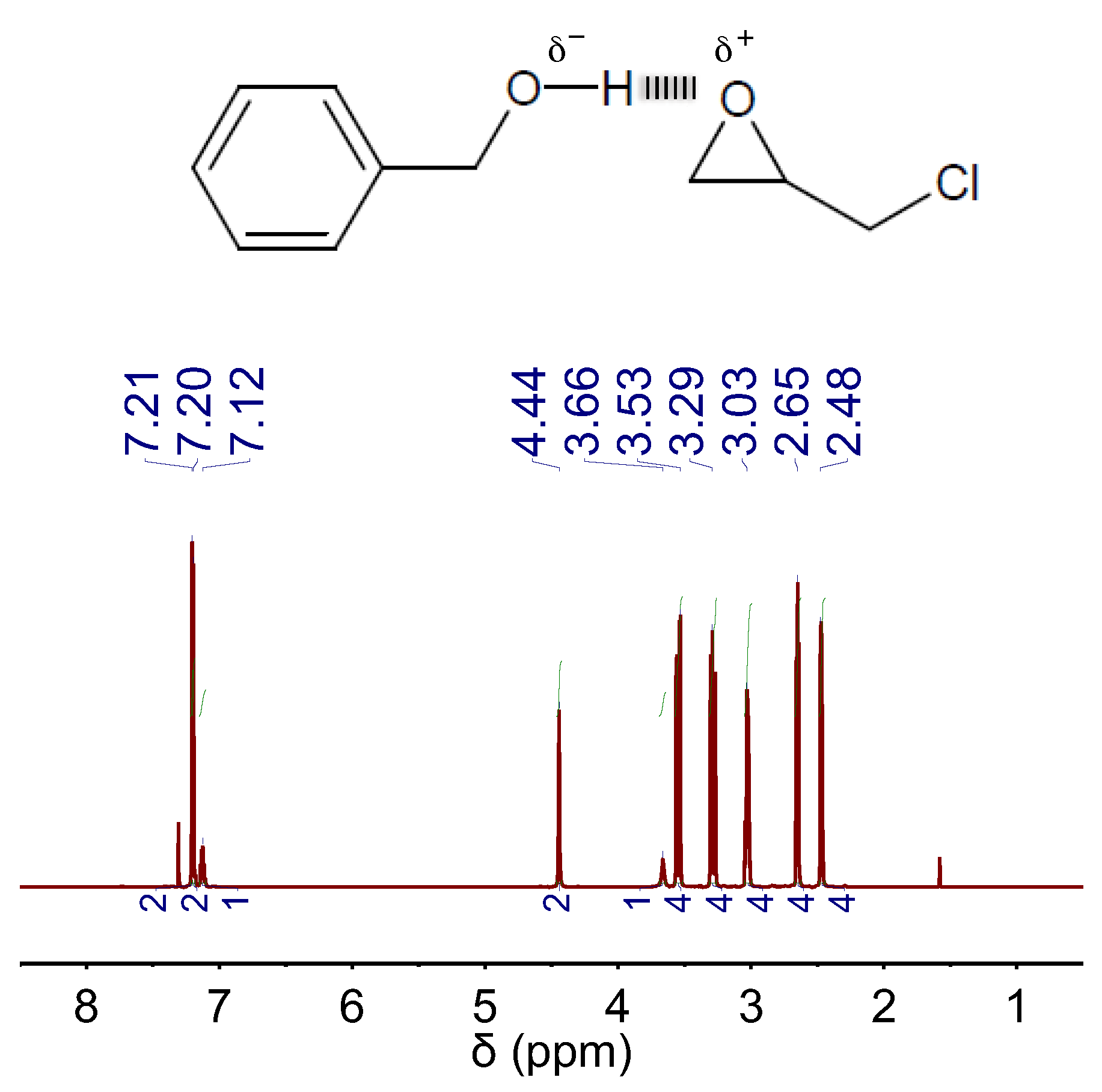
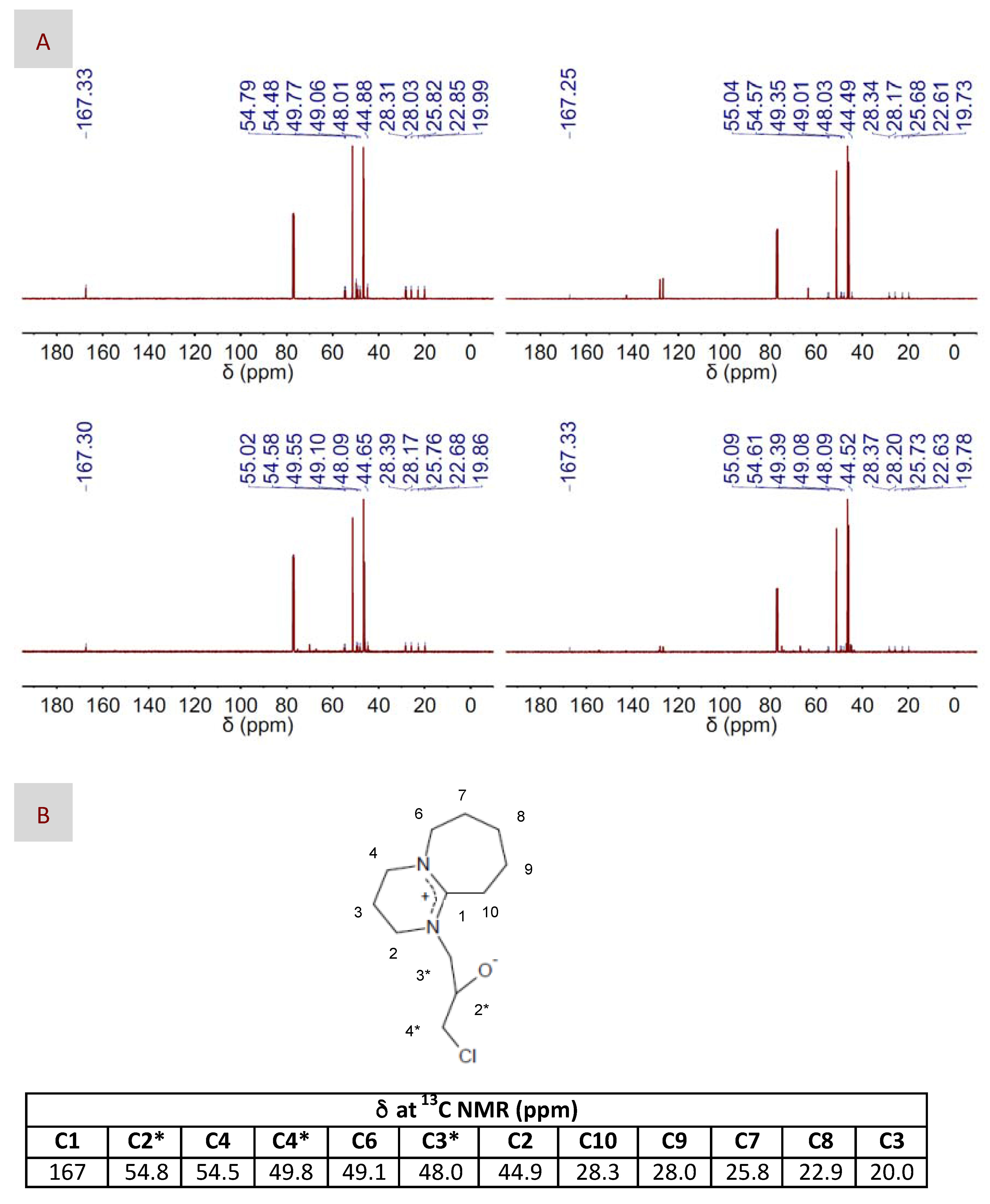
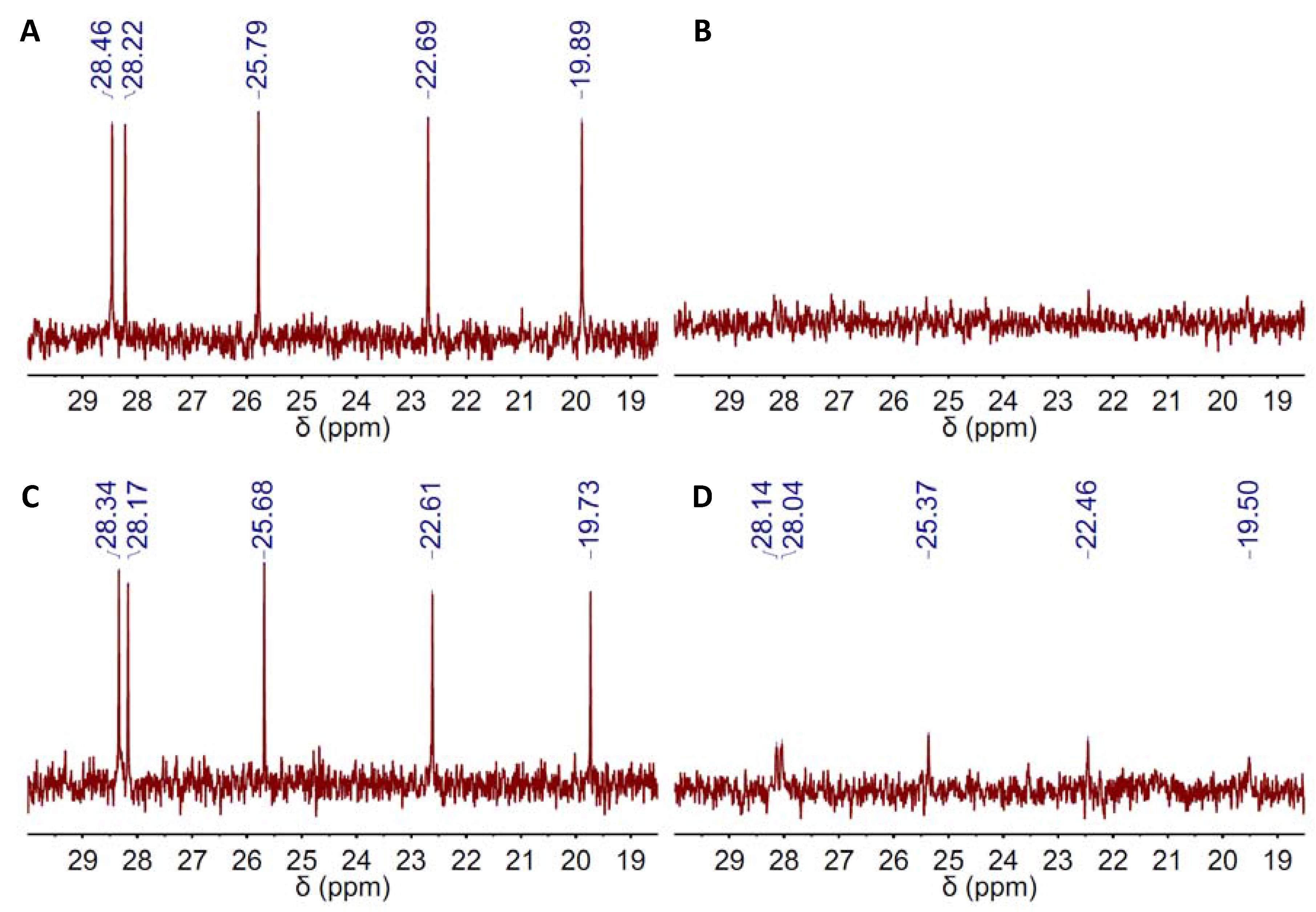
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Preparation of DESs
3.3. CO2 Absorption on DESs
3.4. CO2 Fixation to EP
3.5. Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Williams, C.K.; Hillmyer, M.A. Polymers from renewable resources: A perspective for a special issue of polymer reviews. Polym. Rev. 2008, 48, 1–10. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N. CO2 chemistry: Task-specific ionic liquids for CO2 capture/activation and subsequent conversión. RSC Adv. 2011, 1, 545–567. [Google Scholar] [CrossRef]
- Otto, A.; Grube, T.; Schiebahn, S.; Stolten, D. Closing the loop: Captured CO2 as a feedstock in the chemical industry. Energy Environ. Sci. 2015, 8, 3283–3297. [Google Scholar] [CrossRef]
- International Energy Agency. Available online: http://www.iea.org/newsroomandevents/news/2015/march/global-energy-related-emissions-of-carbon-dioxide-stalled-in-2014.html (accessed on 13 March 2015).
- Andersson, A.M.; Abraham, D.P.; Haasch, R.; MacLaren, S.; Liu, J.; Amine, K. Surface characterization of electrodes from high power lithium-ion batteries. J. Electrochem. Soc. 2002, 149, A1358–A1369. [Google Scholar] [CrossRef]
- Smitha, B.; Sridhar, S.; Khan, A.A. Synthesis and characterization of proton conducting polymer membranes for fuel cells. J. Membr. Sci. 2003, 225, 63–76. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Langanke, J.; Greiner, L.; Leitner, W. Substrate dependent synergetic and antagonistic interaction of ammonium halide and polyoxometalate catalysts in the synthesis of cyclic carbonates from oleo chemical epoxides and CO2. Green Chem. 2013, 15, 1173–1182. [Google Scholar] [CrossRef]
- Song, Q.-W.; He, L.-N.; Wang, J.-Q.; Yasuda, H.; Sakakura, T. Catalytic fixation of CO2 to cyclic carbonates by phosphonium chlorides immobilized on fluorous polymer. Green Chem. 2013, 15, 110–115. [Google Scholar] [CrossRef]
- Yu, K.-M.-K.; Curcic, I.; Gabriel, J.; Morganstewart, H.; Tsang, S.C. Catalytic coupling of CO2 with epoxide over supported and unsupported amines. J. Phys. Chem. A 2010, 114, 3863–3872. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cheng, W.; Yang, Z.; Wang, J.; Xu, T.; Xin, J.; Zhang, S. Superbase/cellulose: An environmentally benign catalyst for chemical fixation of carbon dioxide into cyclic carbonates. Green Chem. 2014, 16, 3071–3078. [Google Scholar] [CrossRef]
- Ema, T.; Fukuhara, K.; Sakai, T.; Ohbo, M.; Bai, F.-Q.; Hasegawa, J.-Y. Quaternary ammonium hydroxide as a metal-free and halogen-free catalyst for the synthesis of cyclic carbonates from epoxides and carbon dioxide. Catal. Sci. Technol. 2015, 5, 2314–2321. [Google Scholar] [CrossRef]
- Roshan, K.R.; Mathai, G.; Kim, J.; Tharun, J.; Park, G.A.; Park, D.W. A biopolymer mediated efficient synthesis of cyclic carbonates from epoxides and carbon dioxide. Green Chem. 2012, 14, 2933–2940. [Google Scholar] [CrossRef]
- Ema, T.; Miyazaki, Y.; Taniguchi, T.; Takada, J. Robust porphyrin catalysts immobilized on biogenous iron oxide for the repetitive conversions of epoxides and CO2 into cyclic carbonates. Green Chem. 2013, 15, 2485–2492. [Google Scholar] [CrossRef]
- Melendez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, Y.M.; Zhang, W.Z.; Qu, J.P.; Lu, X.B. N-Hetero cyclic carbene functionalized MCM-41 as an efficient catalyst for chemical fixation of carbon dioxide. Green Chem. 2011, 13, 644–650. [Google Scholar] [CrossRef]
- Ramidi, P.; Munshi, P.; Gartia, Y.; Pulla, S.; Biris, A.S.; Paul, A.; Ghosh, A. Synergistic effect of alkalihalide and Lewis base on the catalytic synthesis of cyclic carbonate from CO2 and epoxide. Chem. Phys. Lett. 2011, 512, 273–277. [Google Scholar] [CrossRef]
- Cokoja, M.; Wilhelm, M.E.; Anthofer, M.H.; Herrmann, W-A.; Kühn, F.E. Synthesis of cyclic carbonates from epoxides and carbon dioxide by using organocatalysts. ChemSusChem 2015, 8, 2436–2454. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lin, L.; Zhang, G.; Kodama, K.; Yasutake, M.; Hirose, T. Synthesis of cyclic carbonates from CO2 and epoxides catalyzed by low loadings of benzyl bromide/DMF at ambient pressure. Chem. Commun. 2014, 50, 14183–14186. [Google Scholar]
- Liu, X.; Zhang, S.; Song, Q.-W.; Liu, X.-F.; Ma, R.; He, L.-N. Cooperative calcium-based catalysis with 1,8-diazabicyclo[5.4.0]-undec-7-ene for the cycloaddition of epoxides with CO2 at atmospheric pressure. Green Chem. 2016. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Kodama, K.; Hirose, T. Anefficient metal- and solvent-free órgano catalytic system for chemical fixation of CO2 into cyclic carbonates under mild conditions. Green Chem. 2016, 18, 1229–1233. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Sun, J.; Cheng, W.-G.; Dong, K.; Zhang, X.-P.; Zhang, S.-J. Experimental and theoretical studies on hydrogen bond-promoted fixation of carbón dioxide and epoxides in cyclic carbonates. Phys. Chem. Chem. Phys. 2012, 14, 11021–11026. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Han, L.; Cheng, W.; Wang, J.; Zhang, X.; Zhang, S. Efficient Acid–Base Bifunctional Catalysts for the Fixation of CO2 with Epoxides under Metal- and Solvent-Free Conditions. ChemSusChem 2011, 4, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Gennen, S.; Alves, M.; Méreau, R.; Tassaing, T.; Gilbert, B.; Detrembleur, C.; Jerome, C.; Grignard, B. Fluorinated alcohols as activators for the solvent-free chemical fixation of carbon dioxide into epoxides. ChemSusChem 2015, 8, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Xiao, B.; Sun, J.; Dong, K.; Zhang, P.; Zhang, S.; Ng, F.T.T. Effect of hydrogen bond of hydroxyl-functionalized ammonium ionic liquids on cycloaddition of CO2. Tetrahedron Lett. 2015, 56, 1416–1419. [Google Scholar] [CrossRef]
- Tharun, J.; Mathai, G.; Kathalikkattil, A.C.; Roshan, R.; Kwak, J.Y.; Park, D.W. Microwave-assisted synthesis of cyclic carbonates by a formic acid/KI catalytic system. Green Chem. 2013, 15, 1673–1677. [Google Scholar] [CrossRef]
- Xu, B.-H.; Wang, J.-Q.; Sun, J.; Huang, Y.; Zhang, J.-P.; Zhang, X.-P.; Zhang, S.-J. Fixation of CO2 into cyclic carbonates catalyzed by ionic liquids: A multi-scale approach. Green Chem. 2015, 17, 108–122. [Google Scholar] [CrossRef]
- Takenaka, N.; Chen, J.S.; Captain, B.; Sarangthem, R.S.; Chandrakumar, A. Helical chiral 2-aminopyridinium ions: A new class of hydrogen bond donor catalysts. J. Am. Chem. Soc. 2010, 132, 4536–4537. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, B.G.; Young, K.J.H.; Yousufuddin, M.; Goddard, W.A.; Periana, R.A. Acceleration of nucleophilic CH activation by strongly basic solvents. J. Am. Chem. Soc. 2010, 132, 12542–12545. [Google Scholar] [CrossRef] [PubMed]
- Sardon, H.; Engler, A.C.; Chan, J.M.W.; García, J.M.; Coady, D.J.; Pascual, A.; Mecerreyes, D.; Jones, G.O.; Rice, J.E.; Horn, H.W.; et al. Organic acid-cata-lyzed polyurethane formation via a dual-activated mechanism: Unexpected preference of N-Activation over O-Activation of isocyanates. J. Am. Chem. Soc. 2013, 135, 16235–16241. [Google Scholar] [CrossRef] [PubMed]
- Chuma, A.; Horn, H.W.; Swope, W.C.; Pratt, R.C.; Zhang, L.; Lohmeijer, B.G.G.; Wade, C.G.; Waymouth, R.M.; Hedrick, J.L.; Rice, J.E. The reaction mechanism for the organocatalytic ring-opening polymerization of L-Lactide using a guanidine-based catalyst: Hydrogen-bonded or covalently bound. J. Am. Chem. Soc. 2008, 130, 6749–6754. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 70–71. [Google Scholar] [CrossRef]
- Imperato, G.; Eibler, E.; Niedermaier, J.; König, B. Low-melting sugar–urea–salt mixtures as solvents for Diels–Alder reactions. Chem. Commun. 2005, 9, 1170–1172. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; van Spronsen, J.; Dai, Y.; Verberne, M.; Hollmann, F.; Arends, I.W.C.E.; Witkamp, G.-J.; Verpoorte, R. Are natural deep eutectic solvents the missing link in understanding cellular metabolism and physiology? Plant Physiol. 2011, 156, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Francisco, M.; van den Bruinhorst, A.; Kroon, M.C. Low-transition-temperature mixtures (LTTMs): A new generation of designer solvents. Angew. Chem. 2013, 52, 3074–3085. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Leron, R.B.; Li, M.-H. Solubility of carbon dioxide in aqueous mixtures of (reline + monoethanolamine) at T = (313.2 to 353.2) K. J. Chem. Thermodyn. 2014, 72, 94–99. [Google Scholar] [CrossRef]
- Ullah, R.; Atilhan, M.; Anaya, B.; Khraisheh, M.; García, G.; El Khattat, A.; Tariqa, M.; Aparicio, S. A detailed study of cholinium chloride and levulinic acid deep eutectic solvent system for CO2 capture via experimental and molecular simulation approaches. Phys. Chem. Chem. Phys. 2015, 17, 20941–20960. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Leron, R.B.; Li, M.-H. Imidazole tailored deep eutectic solvents for CO2 capture enhanced by hydrogen bonds. Phys. Chem. Chem. Phys. 2015, 17, 27306–27316. [Google Scholar]
- Mirza, N.R.; Nicholas, N.J.; Wu, Y.; Mumford, K.A.; Kentish, S.E.; Stevens, G.W. Experiments and thermodynamic modeling of the solubility of carbon dioxide in three different deep eutectic solvents (DESs). J. Chem. Eng. Data 2015, 60, 3246–3252. [Google Scholar] [CrossRef]
- Altamash, T.; Atilhan, M.; Aliyan, A.; Ullah, R.; García, G.; Aparicio, S. Insights into choline chloride–phenyl acetic acid deep eutectic solvent for CO2 absorption. RSC Adv. 2016, 6, 109201–109210. [Google Scholar] [CrossRef]
- Leron, R.B.; Li, M.-H. Solubility of carbón dioxide in a eutectic mixture of choline chloride and glycerol at moderate pressures. J. Chem. Thermodyn. 2013, 57, 131–136. [Google Scholar] [CrossRef]
- Francisco, M.; van den Bruinhorst, A.; Zubeir, L.F.; Peters, C.J.; Kroon, M.C. A new low transition temperature mixture (LTTM) formed by choline chloride + lactic acid: Characterization as solvent for CO2 capture. Fluid Phase Equilib. 2013, 340, 77–84. [Google Scholar] [CrossRef]
- Trivedi, T.J.; Lee, J.H.; Lee, H.J.; Jeong, Y.K.; Choi, J.W. Deep eutectic solvents as attractive media for CO2 capture. Green Chem. 2016, 18, 2834–2842. [Google Scholar] [CrossRef]
- Ali, E.; Hadj-Kali, M.K.; Mulyono, S.; Alnashef, I. Analysis of operating conditions for CO2 capturing process using deep eutectic solvents. Int. J. Greenhouse Gas Control 2016, 47, 342–350. [Google Scholar] [CrossRef]
- Liu, M.; Lia, X.; Lina, X.; Liang, L.; Gao, X.; Sun, J. Facile synthesis of [urea-Zn]I2 eutectic-based ionic liquid for efficient conversion of carbon dioxide to cyclic carbonates. J. Mol. Catal. A-Chem. 2016, 412, 20–26. [Google Scholar] [CrossRef]
- Saptal, V.B.; Bhanage, B.M. Bifunctional ionic liquids derived from biorenewable sources as sustainable catalysts for fixation of carbon dioxide. ChemSusChem 2016, 10, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Carriazo, D.; Serrano, M.C.; Gutiérrez, M.C.; Ferrer, M.L.; Del Monte, F. Deep eutectic solvents in polymerizations: A greener alternative to conventional syntheses. ChemSusChem 2014, 7, 999–1009. [Google Scholar]
- Sze, L.L.; Pandey, S.; Ravula, S.; Pandey, S.; Zhao, H.; Baker, G.A.; Baker, S.N. Ternary deep eutectic solvents tasked for carbon dioxide capture. ACS Sustain. Chem. Eng. 2014, 2, 2117–2123. [Google Scholar] [CrossRef]
- Sarmad, S.; Mikkola, J.-P.; Ji, X. Carbon dioxide capture with ionic liquids and deep eutectic solvents: A new generation of sorbents. ChemSusChem 2017, 10, 324–352. [Google Scholar] [CrossRef] [PubMed]
- García-Argüelles, S.; García, C.; Serrano, M.C.; Gutiérrez, M.C.; Ferrer, M.L.; Del Monte, F. Near-to-eutectic mixtures as bifunctional catalysts in the low-temperature-ring-opening-polymerization of ε-caprolactone. Green Chem. 2015, 17, 3632–3643. [Google Scholar] [CrossRef]
- Coady, D.J.; Horn, H.W.; Jones, G.O.; Sardon, H.; Engler, A.C.; Waymouth, R.M.; Rice, J.E.; Yang, Y.Y.; Hedrick, J.L. Polymerizing base sensitive cyclic carbonates using acid catalysis. ACS Macrolett. 2013, 2, 306–312. [Google Scholar] [CrossRef]
- Guillerm, B.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Dubois, P.; Coulembier, O. Ammonium betaines: Efficient ionic nucleophilic catalysts for the ring-opening polymerization of L-lactide and cyclic carbonates. Chem Commun. 2014, 50, 10098–10101. [Google Scholar] [CrossRef] [PubMed]
- Heldebrant, D.J.; Yonker, C.R.; Jessop, P.G.; Phan, L. Organic liquid CO2 capture agents with high gravimetric CO2 capacity. Energy Environ. Sci. 2008, 1, 487–493. [Google Scholar] [CrossRef]
- Wang, C.; Mahurin, S.M.; Luo, H.; Baker, G.A.; Li, H.; Dai, S. Reversible and robust CO2 capture by equimolar task-specific ionic liquid–superbase mixtures. Green Chem. 2010, 12, 870–874. [Google Scholar] [CrossRef]
- Wang, C.; Luo, H.; Luo, X.; Li, H.; Dai, S. Equimolar CO2 capture by imidazolium-based ionic liquids and superbase systems. Green Chem. 2010, 12, 2019–2023. [Google Scholar] [CrossRef]
- Taylor, S.F.R.; McCrellis, C.; McStay, C.; Jacquemin, J.; Hardacre, C.; Mercy, M.; Bell, R.G.; De Leeuw, N.H. CO2 Capture in wet and dry superbase ionic liquids. J. Solution Chem. 2015, 44, 511–527. [Google Scholar] [CrossRef]
- Taylor, S.F.R.; McCrellis, C.; McStay, C.; Jacquemin, J.; Hardacre, C.; Mercy, M.; Bell, R.G.; De Leeuw, N.H. Efficient fixation and conversion of CO2 into dimethyl carbonate catalyzed by animidazolium containing tri-cationic ionic liquid/super base system. RSC Adv. 2016, 4, 42279–42287. [Google Scholar]
- Yang, Z.-Z.; He, L.-N.; Miao, C.-X.; Chanfreau, S. Lewis basic ionic liquids-catalyzed conversion of carbon dioxide to cyclic carbonates. Adv. Synth. Catal. 2010, 352, 2233–2240. [Google Scholar] [CrossRef]
- Foltran, S.; Alsarraf, J.; Robert, F.; Landais, Y.; Cloutet, E.; Cramail, H.; Tassaing, T. On the chemical fixation of super critical carbón dioxide with epoxides catalyzed by ionic salts: An in situ FTIR and Raman study. Catal. Sci. Technol. 2013, 3, 1046–1055. [Google Scholar] [CrossRef]
- Coulembier, O.; Moins, S.; Lemaur, V.; Lazzaroni, R.; Dubois, P. Efficiency of DBU/iodine cooperative dual catalysis for the solvent-free synthesis of five-membered cyclic carbonates under atmospheric CO2 pressure. J. CO2 Util. 2015, 10, 7–11. [Google Scholar] [CrossRef]
- Wang, L.; Kodama, K.; Hirose, T. DBU/benzylbromide: An efficient catalytic system for the chemical fixation of CO2 into cyclic carbonates under metal- and solvent- free conditions. Catal. Sci. Technol. 2016, 6, 3872–3877. [Google Scholar] [CrossRef]
- Wang, C.; Luo, H.; Jiang, D.-E.; Li, H.; Dai, S. Carbon dioxide capture by superbase-derived protic ionic liquids. Angew. Chem. 2010, 49, 5978–5981. [Google Scholar] [CrossRef] [PubMed]
- Jessop, P.G.; Mercer, S.M.; Heldebrant, D.J. CO2-triggered switchable solvents, surfactants, and other materials. Energy Environ. Sci. 2012, 5, 7240–7253. [Google Scholar] [CrossRef]
- Pérez, E.R.; Santos, R.H.A.; Gambardella, M.T.P.; De Macedo, L.G.M.; Rodrigues-Filho, U.P.; Launay, J.-C.; Franco, D.W. Activation of carbon dioxide by bicyclic amidines. J. Org. Chem. 2004, 69, 8005–8011. [Google Scholar] [CrossRef] [PubMed]
- Heldebrant, D.J.; Jessop, P.G.; Thomas, C.A.; Eckert, C.A.; Liotta, C.L. The Reaction of 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) with carbon dioxide. J. Org. Chem. 2005, 70, 5335–5338. [Google Scholar] [CrossRef] [PubMed]
- Anthofer, M.H.; Wilhelm, M.E.; Cokoja, M.; Markovits, I.I.E.; Pöthig, A.; Mink, J.; Herrmann, W.A.; Kühn, F.E. Cycloaddition of CO2 and epoxides catalyzed by imidazolium bromides under mild conditions: Influence of the cation on catalyst activity. Catal. Sci. Technol. 2014, 4, 1749–1758. [Google Scholar] [CrossRef]
- Hardman-Baldwin, A.M.; Mattson, A.E. Silanediol-catalyzed carbon dioxide fixation. ChemSusChem 2014, 7, 3275–3278. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DES | mol CO2/mol Superbase |
|---|---|
| DBU(1):BA(1) | 0.28–0.25 |
| DBU(1):BA(4) | 0.90–1 |
| DBU(1):EG(1) | 0.33–0.3 |
| DBU(1):EG(4) | 0.98–1 |
| DBU(1):MDEA(2) | 0.50–0.5 |
| TBD(1):BA(1) | 0.45–0.5 |
| TBD(1):BA(4) | 0.96–1 |
| TBD(1):EG(1) | 0.50–0.5 |
| TBD(1):EG(4) | 1–1 |
| TBD(1):MDEA(2) | 0.52–0.5 |
| Entry | Type of Catalyst | Molar Ratio of Catalyst Versus Reagent | T (°C) | t (h) | P (bars) | Yield (%) | Selectivity (%) |
|---|---|---|---|---|---|---|---|
| #1 | DBU | 1:100 | 100 | 1 | 6 | 60 a/64 b | 98 a/98 b |
| #2 | DBU | 1:100 | 100 | 2 | 6 | 82 a/88 b | 98 a/98 b |
| #3 | DBU | 1:100 | 100 | 1 | 1.2 | 72 a/81 b | 99 a/99 b |
| #4 | DBU | 1:100 | 100 | 2 | 1.2 | 84 a/87 b | 99 a/99 b |
| #5 | DBU | 1:100 | 50 | 2 | 1.2 | 27 a/9 b | 90 a/90 b |
| #6 | DBU | 1:100 | 50 | 20 | 1.2 | 49 a/52 b | 97 a/97 b |
| #7 | DBU | 10:100 | 50 | 2 | 1.2 | 42 a/43 b | 75 a/75 b |
| #8 | DBU | 10:100 | 50 | 20 | 1.2 | 92 a/95 b | 98 a/98 b |
| #9 | TBD | 1:100 | 100 | 2 | 1.2 | 93 a/88 b | 99 a/99 b |
| #10 | TBD | 1:100 | 50 | 2 | 1.2 | 8 a/10 b | 88 a/88 b |
| #11 | TBD | 1:100 | 50 | 20 | 1.2 | 50 a/58 b | 99 a/99 b |
| #12 | TBD | 10:100 | 50 | 2 | 1.2 | 35 a/41 b | 86 a/86 b |
| #13 | TBD | 10:100 | 50 | 20 | 1.2 | 97 a/99 b | 97 a/97 b |
| #14 | DBU(1):BA(1) | 1:100 | 100 | 1 | 6 | 67 a/62 b | 98 a/98 b |
| #15 | DBU(1):BA(1) | 1:100 | 100 | 2 | 6 | 83 a/85 b | 99 a/99 b |
| #16 | DBU(1):BA(1) | 1:100 | 100 | 1 | 1.2 | 73 a/70 b | 98 a/98 b |
| #17 | DBU(1):BA(1) | 1:100 | 100 | 2 | 1.2 | 86 a/93 b | 99 a/99 b |
| #18 | DBU(1):EG(1) | 1:100 | 100 | 1 | 6 | 67 a/74 b | 98 a/98 b |
| #19 | DBU(1):EG(1) | 1:100 | 100 | 2 | 6 | 84 a/86 b | 99 a/99 b |
| #20 | DBU(1):EG(1) | 1:100 | 100 | 1 | 1.2 | 73 a/66 b | 98 a/98 b |
| #21 | DBU(1):EG(1) | 1:100 | 100 | 2 | 1.2 | 83 a/86 b | 98 a/98 b |
| #22 | DBU(1):MDEA(2) | 1:100 | 100 | 1 | 6 | 79 a/79 b | 97 a/97 b |
| #23 | DBU(1):MDEA(2) | 1:100 | 100 | 2 | 6 | 88 a/90 b | 97 a/97 b |
| #24 | DBU(1):MDEA(2) | 1:100 | 100 | 1 | 1.2 | 82 a/81 b | 96 a/96 b |
| #25 | DBU(1):MDEA(2) | 1:100 | 100 | 2 | 1.2 | 91 a/93 b | 97 a/97 b |
| #26 | TBD(1):BA(1) | 1:100 | 100 | 2 | 1.2 | 98 a/96 b | 98 a/98 b |
| #27 | TBD(1):EG(1) | 1:100 | 100 | 2 | 1.2 | 94 a/87 b | 98 a/98 b |
| #28 | TBD(1):MDEA(2) | 1:100 | 100 | 2 | 1.2 | 90 a/97 b | 97 a/97 b |
| #29 | DBU(1):BA(1) | 1:100 | 50 | 2 | 1.2 | 11 a/12 b | 87 a/87 b |
| #30 | DBU(1):BA(1) | 1:100 | 50 | 20 | 1.2 | 58 a/62 b | 98 a/98 b |
| #31 | DBU(1):BA(1) | 10:100 | 50 | 2 | 1.2 | 57 a/57 b | 96 a/96 b |
| #32 | DBU(1):BA(1) | 10:100 | 50 | 20 | 1.2 | 89 a/94 b | 98 a/98 b |
| #33 | TBD(1):BA(1) | 1:100 | 50 | 2 | 1.2 | 8 a/6 b | 88 a/88 b |
| #34 | TBD(1):BA(1) | 1:100 | 50 | 20 | 1.2 | 44 a/46 b | 97 a/97 b |
| #35 | TBD(1):BA(1) | 10:100 | 50 | 2 | 1.2 | 6 a/21 b | 76 a/76 b |
| #36 | TBD(1):BA(1) | 10:100 | 50 | 20 | 1.2 | 93 a/86 b | 93 a/93 b |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Argüelles, S.; Ferrer, M.L.; Iglesias, M.; Del Monte, F.; Gutiérrez, M.C. Study of Superbase-Based Deep Eutectic Solvents as the Catalyst in the Chemical Fixation of CO2 into Cyclic Carbonates under Mild Conditions. Materials 2017, 10, 759. https://doi.org/10.3390/ma10070759
García-Argüelles S, Ferrer ML, Iglesias M, Del Monte F, Gutiérrez MC. Study of Superbase-Based Deep Eutectic Solvents as the Catalyst in the Chemical Fixation of CO2 into Cyclic Carbonates under Mild Conditions. Materials. 2017; 10(7):759. https://doi.org/10.3390/ma10070759
Chicago/Turabian StyleGarcía-Argüelles, Sara, Maria Luisa Ferrer, Marta Iglesias, Francisco Del Monte, and María Concepción Gutiérrez. 2017. "Study of Superbase-Based Deep Eutectic Solvents as the Catalyst in the Chemical Fixation of CO2 into Cyclic Carbonates under Mild Conditions" Materials 10, no. 7: 759. https://doi.org/10.3390/ma10070759
APA StyleGarcía-Argüelles, S., Ferrer, M. L., Iglesias, M., Del Monte, F., & Gutiérrez, M. C. (2017). Study of Superbase-Based Deep Eutectic Solvents as the Catalyst in the Chemical Fixation of CO2 into Cyclic Carbonates under Mild Conditions. Materials, 10(7), 759. https://doi.org/10.3390/ma10070759