In Situ DRIFTS Studies of NH3-SCR Mechanism over V2O5-CeO2/TiO2-ZrO2 Catalysts for Selective Catalytic Reduction of NOx

Abstract
:
1. Introduction
2. Results and Discussion
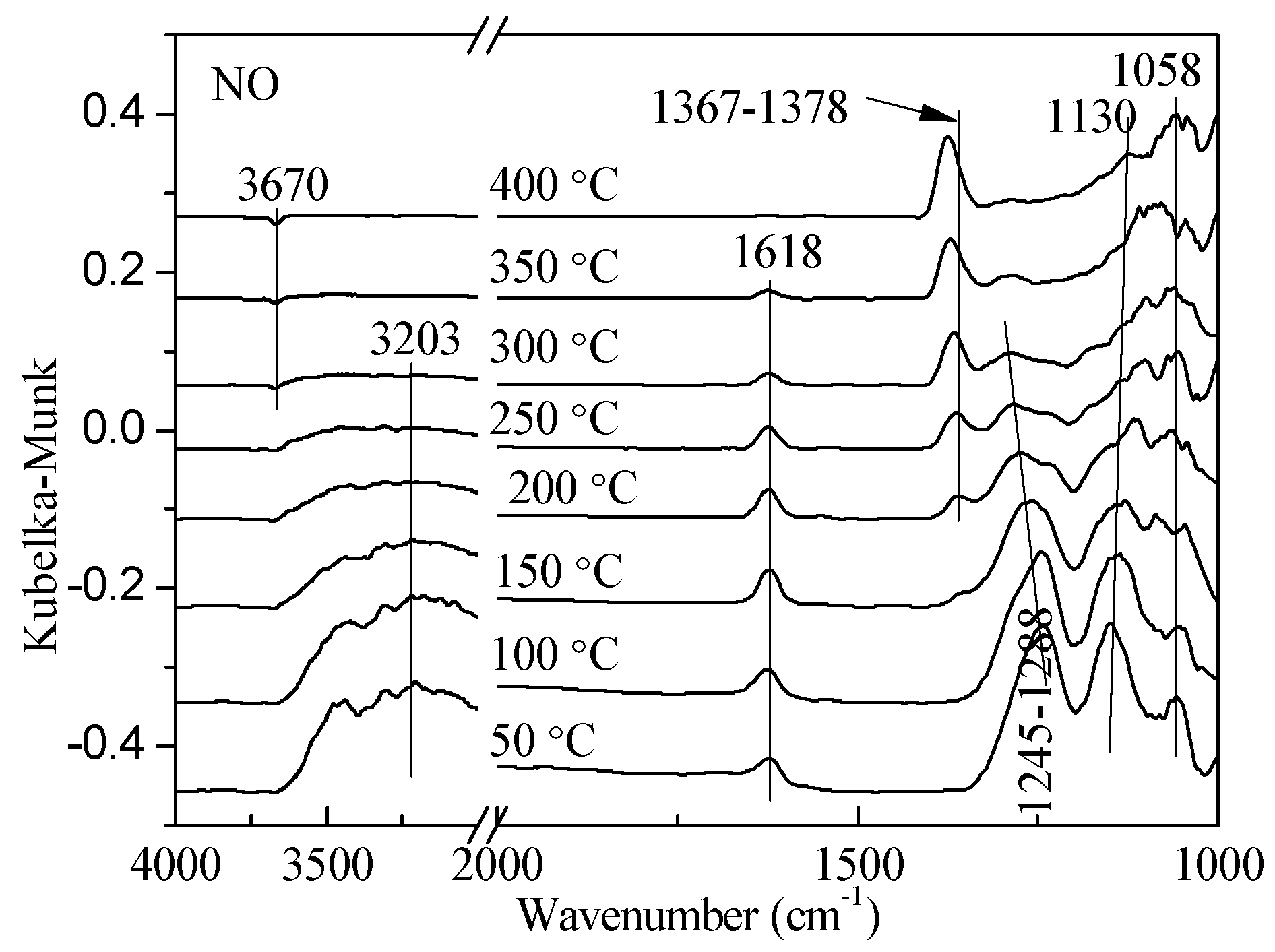
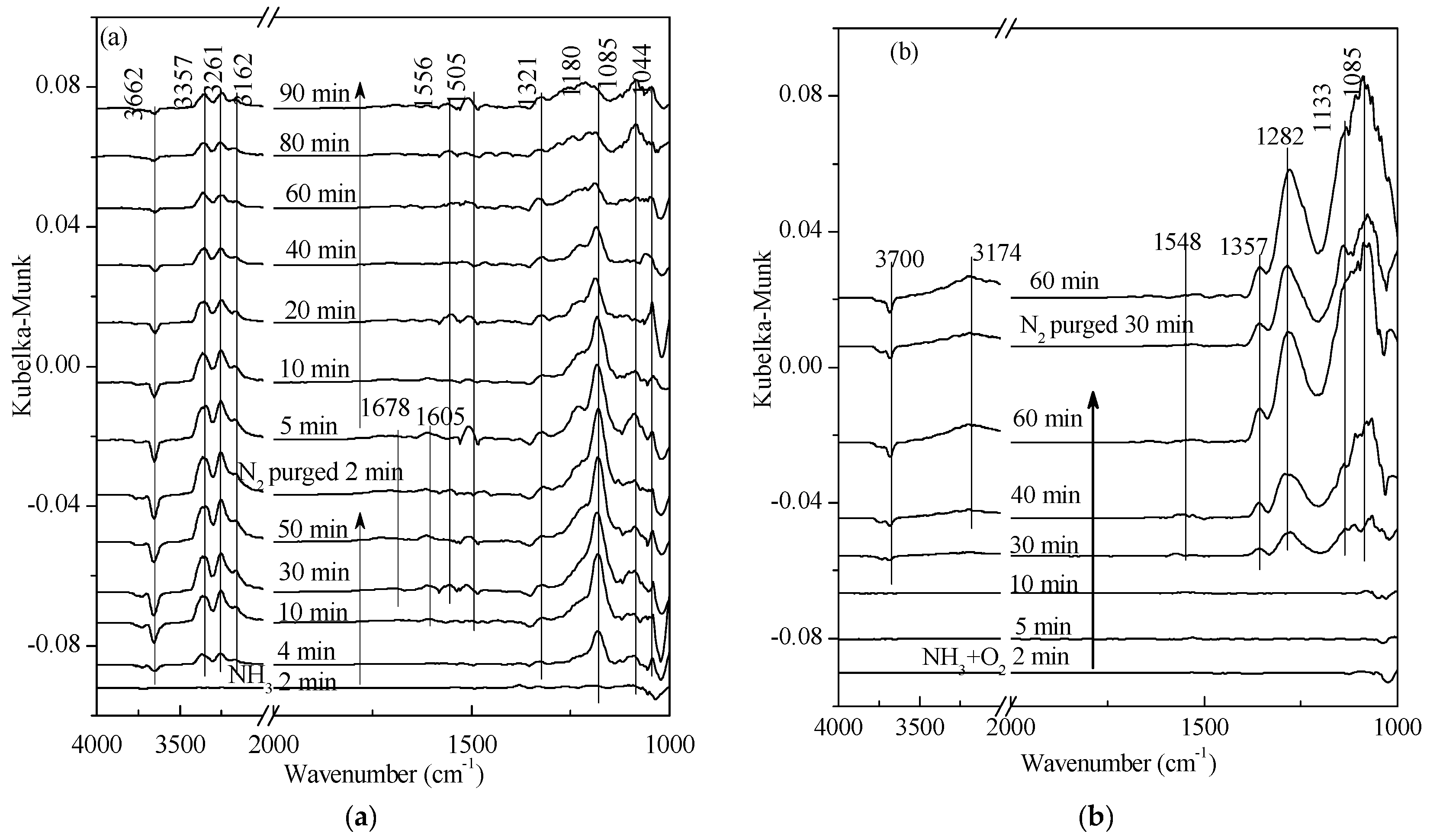
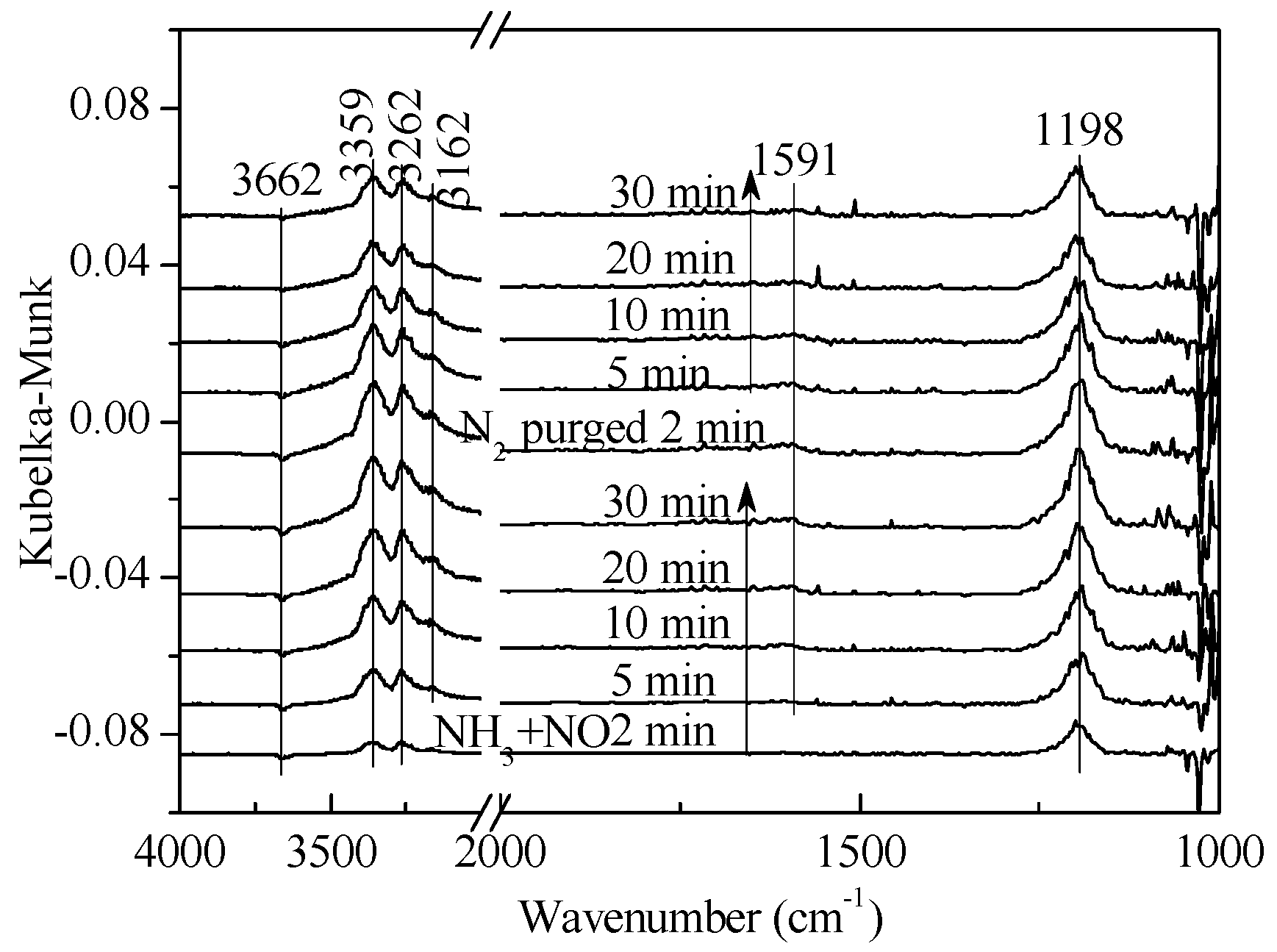
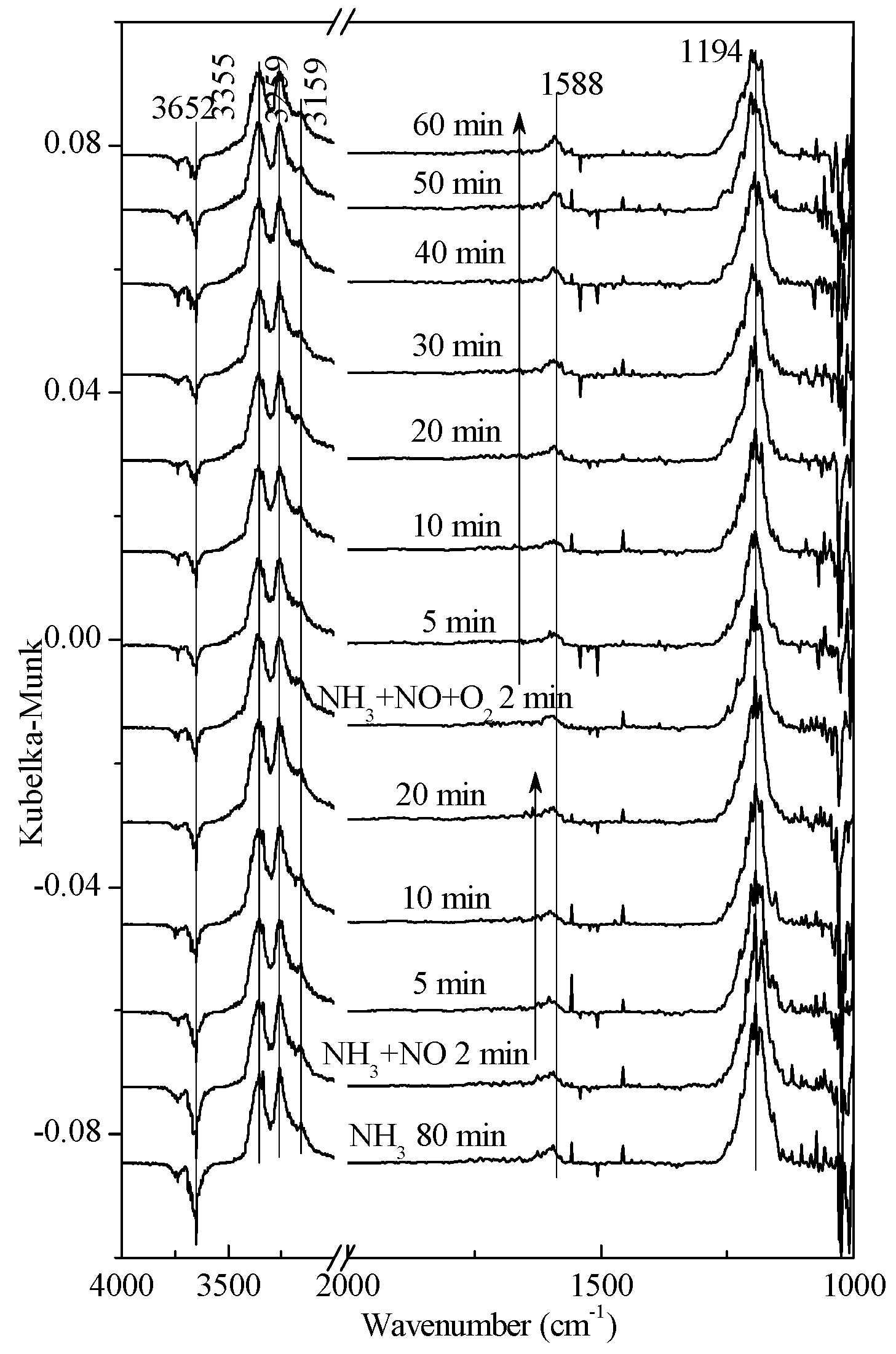
2.1. Adsorption and Desorption Properties of NOx and NH3 on the Catalysts
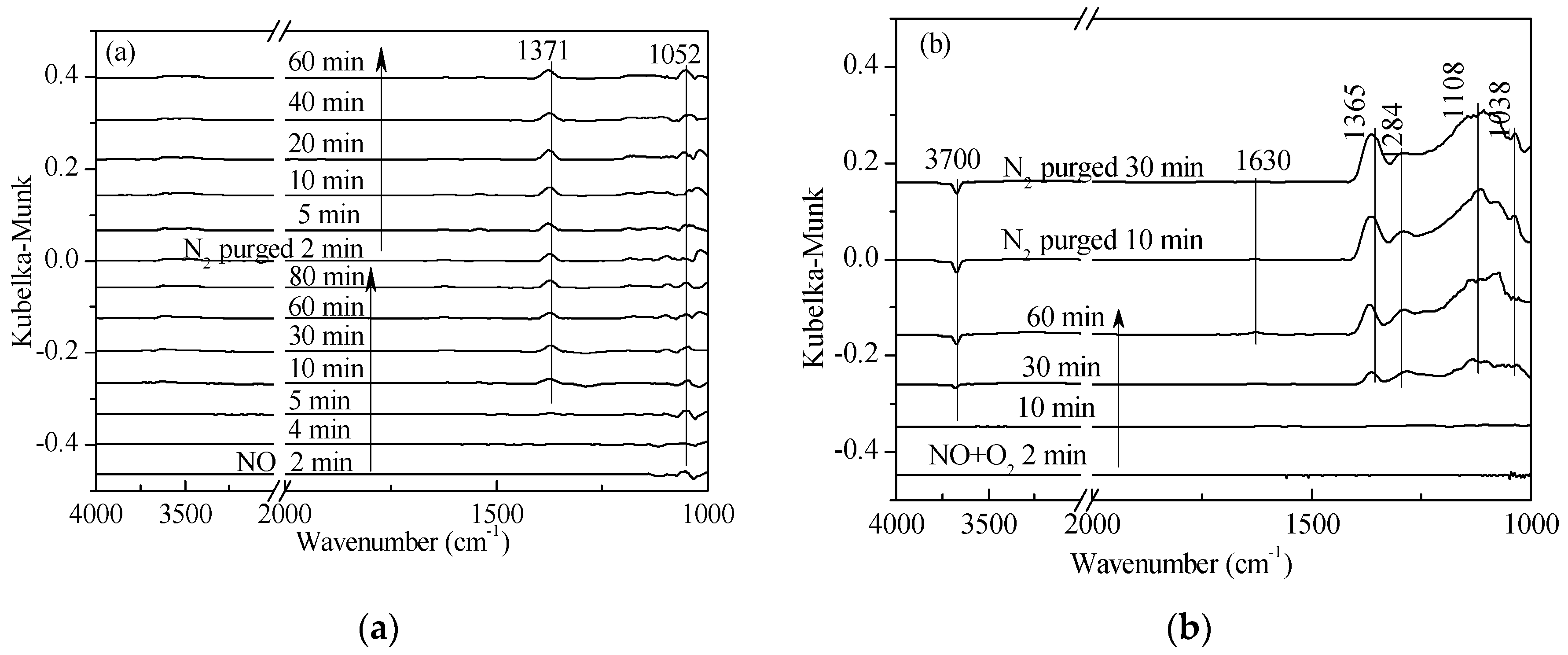
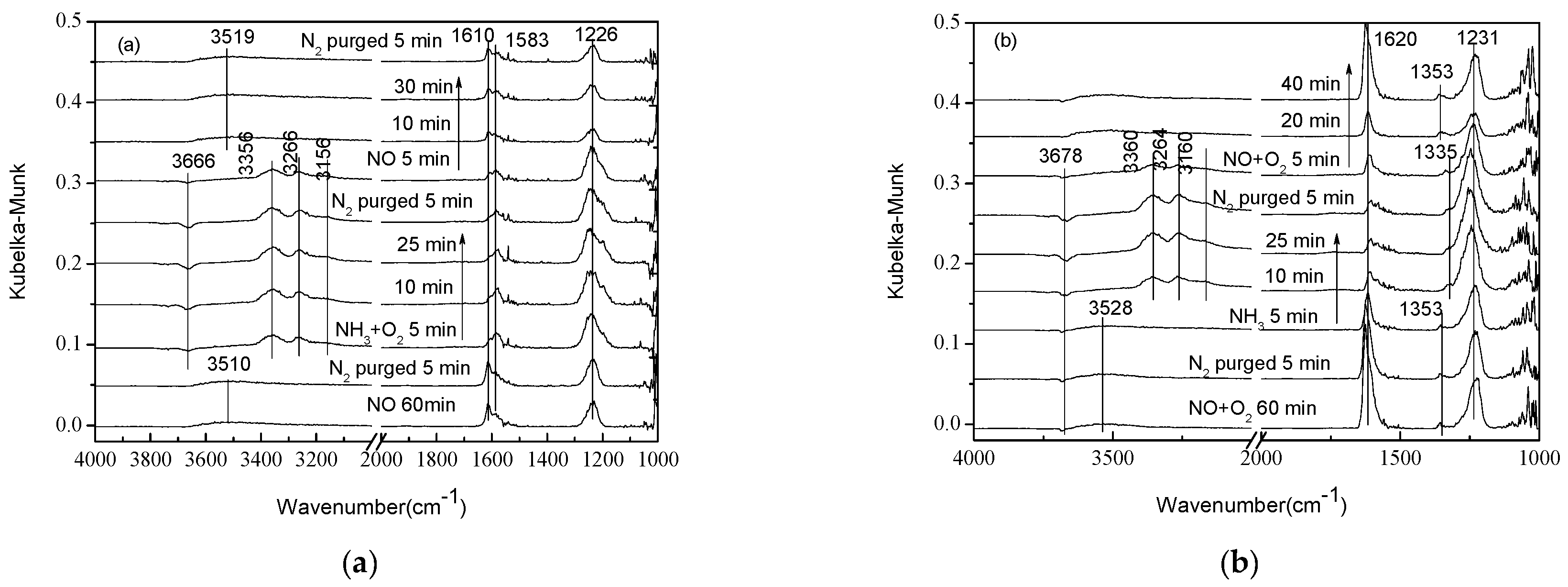
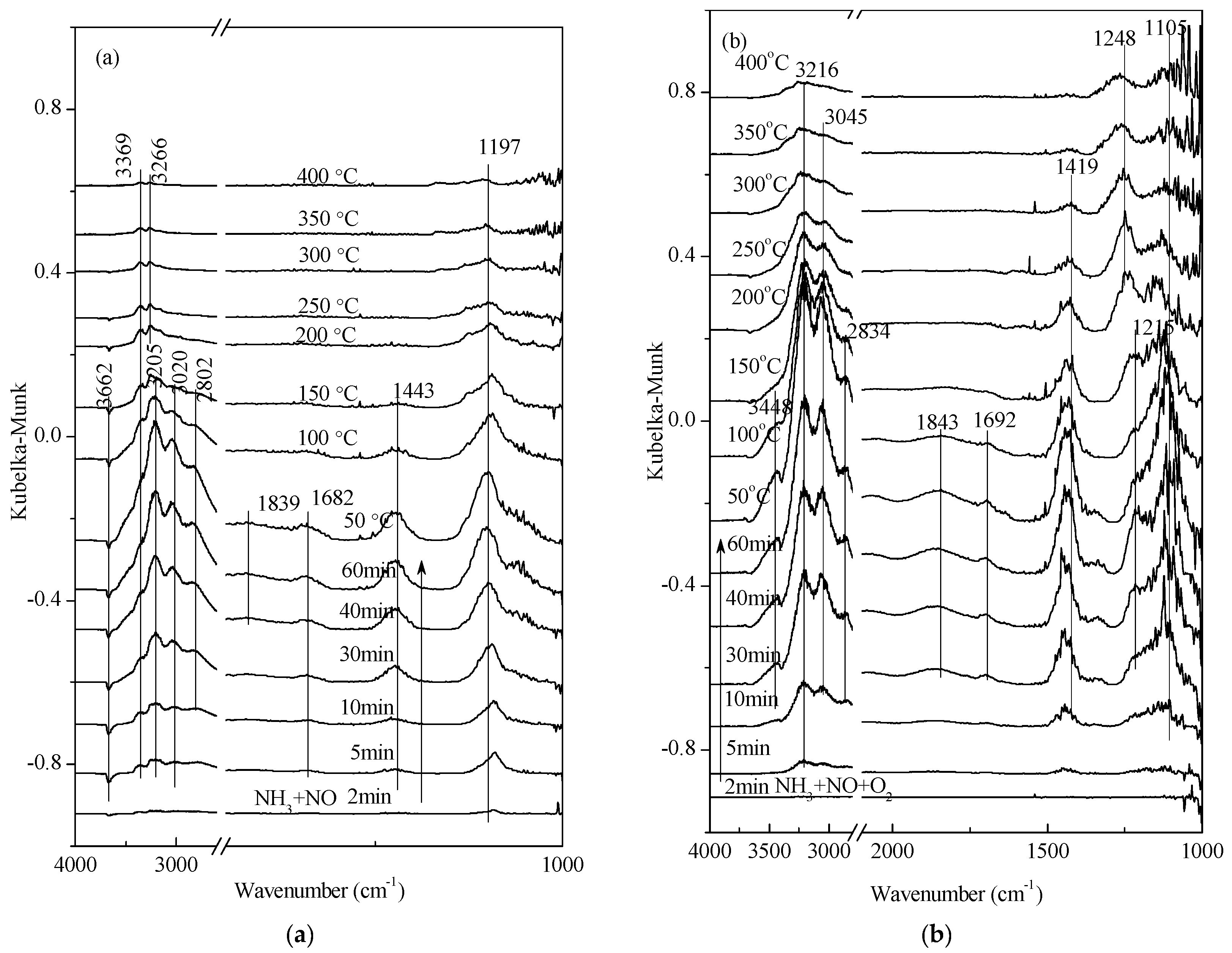
2.2. Transient Response Experiment Analysis
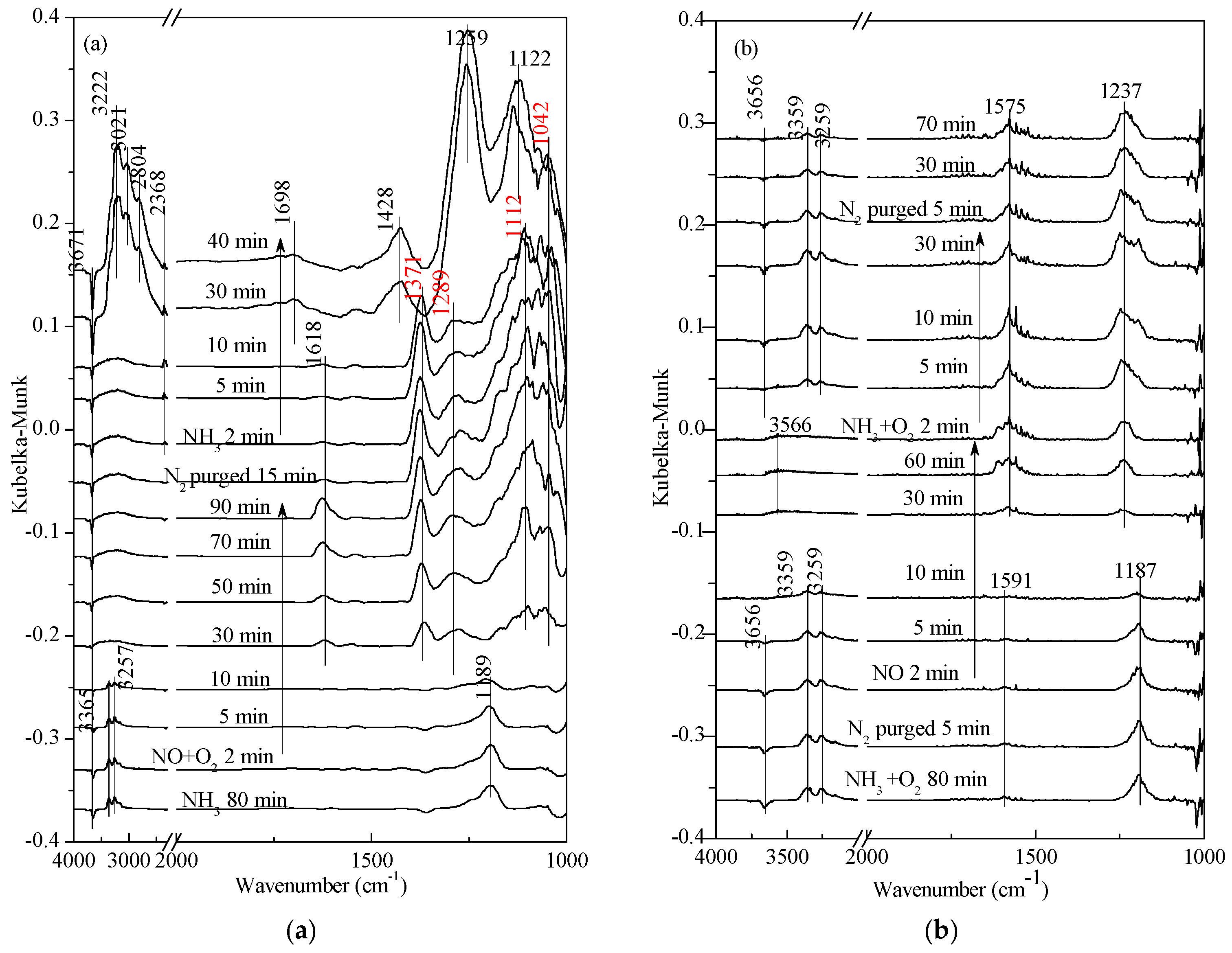
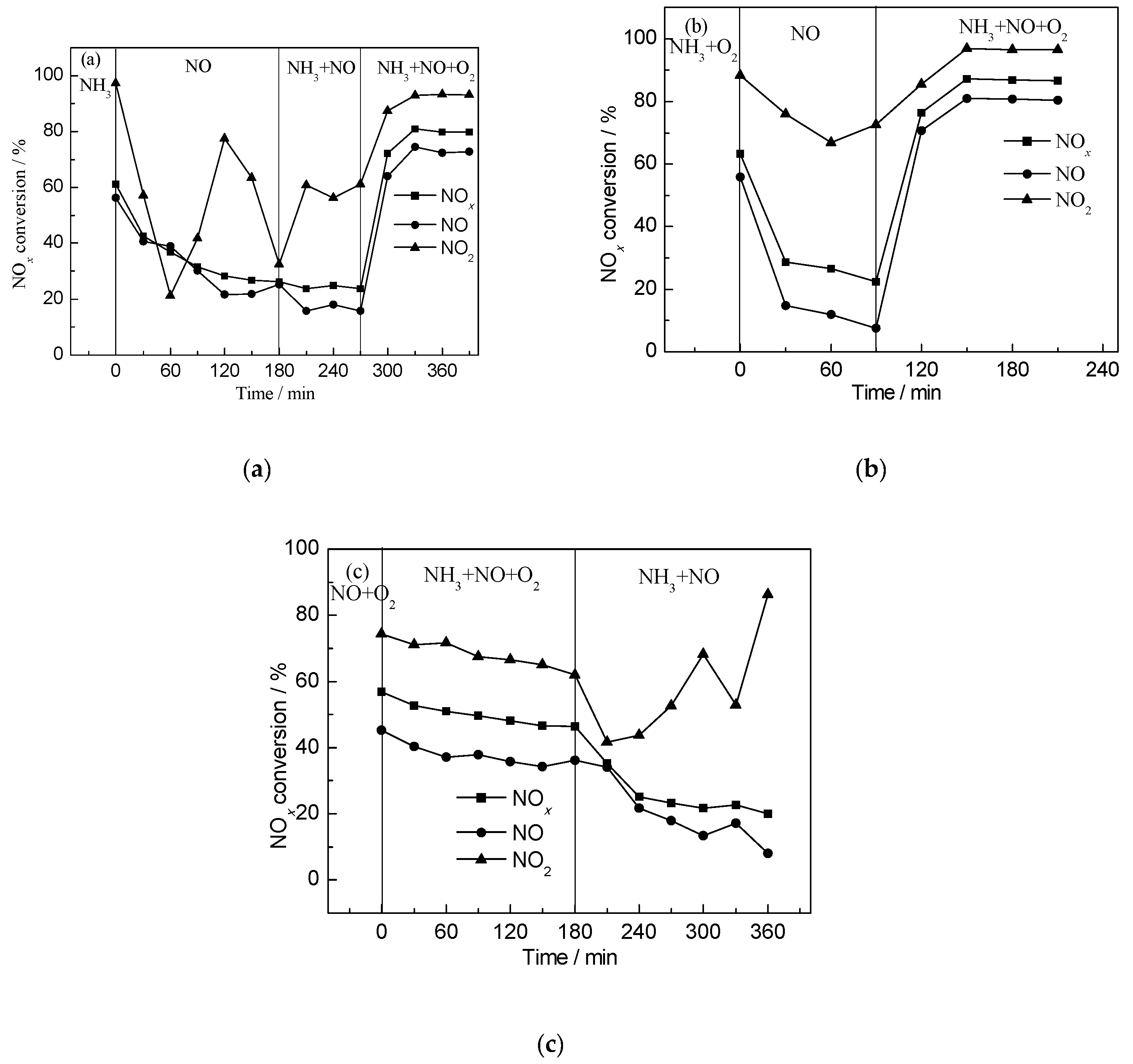
2.3. Steady-State Response Experiments
2.4. Transient SCR Activity Test Experiments
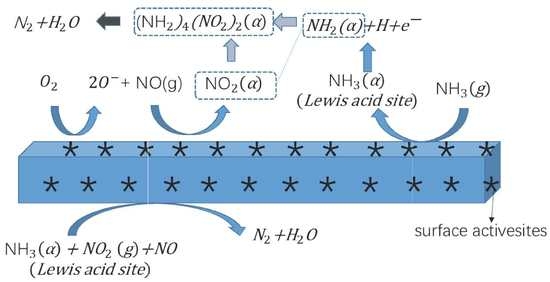
2.5. Low-Temperature SCR Reaction Pathway
3. Materials and Methods
3.1. Catalyst Preparation
3.2. In situ DRIFTS Experiments
3.3. Transient SCR Activity Tests
4. Conclusions
- (1)
- NH3 held a dominant position in the competitive adsorption between NH3 and NO. Transient SCR activity tests showed that the NH3 pre-adsorbed catalyst exhibited better SCR activity than its NOx pre-adsorbed counterpart.
- (2)
- NO might be adsorbed on the catalyst surface and be converted to monodentate nitrite and nitrate species, which is more obvious in the presence of O2, and dramatically restrains the adsorption of NH3, hindering the SCR reaction.
- (3)
- More acid sites and reaction intermediate species -NH2 at lower temperatures mainly led to the higher activity of the V2O5-0.2CeO2/TiO2-ZrO2 catalyst.
- (4)
- Transient SCR activity tests and steady-state response experiments both confirmed that NH3-SCR activity was enhanced by the presence of O2. NH3 adsorption intensity had no obvious difference, whether NO or O2 was introduced or not, indicating that the adsorption and consumption of NH3 was in dynamic equilibrium, which promoted SCR reaction.
- (5)
- NH3-SCR reaction over 1 wt. % V2O5-0.2CeO2/TiO2-ZrO2 catalyst mainly follows the E-R mechanism.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nova, I.; Ciardelli, C.; Tronconi, E.; Chatterjee, D.; Bandlkonrad, B. NH3-SCR of NO over a V-based catalyst: Low-T redox kinetics with NH3 inhibition. AIChE J. 2006, 52, 3222. [Google Scholar] [CrossRef]
- Xu, H.; Shen, Y.; Shao, C.; Lin, F.; Zhu, S.; Qiu, T. A novel catalyst of silicon cerium complex oxides for selective catalytic reduction of NO by NH3. J. Rare Earths 2010, 28, 721–726. [Google Scholar] [CrossRef]
- Topsoe, N.Y.; Dumesic, J.A.; Topsoe, H. Vanadia-titania catalysts for selective catalytic reduction of nitric-oxide by ammonia: I.I. Studies of active sites and formulation of catalytic cycles. J. Catal. 1995, 151, 241–252. [Google Scholar] [CrossRef]
- Arnarson, L.; Falsig, H.; Rasmussen, S.B.; Lauritsen, J.V.; Moses, P.G. The reaction mechanism for the SCR process on monomer V5+ sites and the effect of modified Bronsted acidity. Phys. Chem. Chem. Phys. 2016, 18, 17071–17080. [Google Scholar] [CrossRef] [PubMed]
- Marberger, A.; Ferri, D.; Elsener, M.; Krocher, O. The Significance of Lewis Acid Sites for the Selective Catalytic Reduction of Nitric Oxide on Vanadium-Based Catalysts. Angew. Chem. Int. Ed. 2016, 128, 11989–11994. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Liao, Y.; Dang, H.; Qi, F.; Yang, S. Promotion mechanism of CeO2 addition on the low temperature SCR reaction over MnOx/TiO2: A new insight from the kinetic study. RSC Adv. 2015, 5, 27785–27793. [Google Scholar] [CrossRef]
- Vuong, T.H.; Radnik, J.; Rabeah, J.; Bentrup, U.; Schneider, M.; Atia, H.; Armbruster, U.; Grunert, W.; Bruckner, A. Efficient VOx/Ce1−xTixO2 catalysts for low-temperature NH3-SCR: Reaction mechanism and active sites assessed by in situ/operando spectroscop. ACS Catal. 2017, 7, 1693–1705. [Google Scholar] [CrossRef]
- Zhang, T.; Qu, R.; Su, W.; Li, J. A novel Ce–Ta mixed oxide catalyst for the selective catalytic reduction of NOx with NH3. Appl. Catal. B Environ. 2015, 176, 338–346. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, X.; Shen, K.; Xu, H.; Sun, K.; Zhou, C. Influence of ceria modification on the properties of TiO2–ZrO2 supported V2O5 catalysts for selective catalytic reduction of NO by NH3. J. Colloid Interface Sci. 2012, 376, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, J.; Li, J.; Ma, L.; Woo, S.I. Novel Mn–Ce–Ti Mixed-Oxide catalyst for the selective catalytic reduction of NOx with NH3. ACS Appl. Mater. Interfaces 2014, 6, 14500–14508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pierce, J.; Leung, V.L.; Wang, D.; Epling, W.S. Characterization of ceria’s interaction with NOx and NH3. J. Phys. Chem. C 2013, 117, 8282–8289. [Google Scholar] [CrossRef]
- Galvez, M.E.; Boyano, A.; Lazaro, M.J.; Moliner, R. A study of the mechanisms of NO reduction over vanadium loaded activated carbon catalysts. Chem. Eng. J. 2008, 144, 10–20. [Google Scholar] [CrossRef]
- Yu, J.; Si, Z.; Chen, L.; Wu, X.; Weng, D. Selective catalytic reduction of NOx by ammonia over phosphate-containing Ce0.75Zr0.25O2 solids. Appl. Catal. B Environ. 2015, 163, 223–232. [Google Scholar] [CrossRef]
- Ma, Z.; Wu, X.; Si, Z.; Weng, D.; Ma, J.; Xu, T. Impacts of niobia loading on active sites and surface acidity in NbOx/CeO2–ZrO2 NH3–SCR catalysts. Appl. Catal. B Environ. 2015, 179, 380–394. [Google Scholar] [CrossRef]
- Hu, H.; Zha, K.; Li, H.; Shi, L.; Zhang, D. In situ DRIFTs investigation of the reaction mechanism over MnOx-MOy/Ce0.75Zr0.25O2 (M=Fe, Co, Ni, Cu) for the selective catalytic reduction of NOx with NH3. Appl. Surf. Sci. 2016, 387, 921–928. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, W.; Wang, L.; Song, M.; Yang, L.; Shen, K.; Xu, H.; Zhou, C. Characterization and activity of V2O5-CeO2/TiO2-ZrO2 catalysts for NH3-selective catalytic reduction of NOx. Chin. J. Catal. 2015, 36, 1701–1710. [Google Scholar] [CrossRef]
- Underwood, G.M.; Miller, T.M.; Grassian, V.H. Transmission FT-IR and knudsen cell study of the heterogeneous reactivity of gaseous nitrogen dioxide on mineral oxide particles. J. Phys. Chem. A 1999, 103, 6184–6190. [Google Scholar] [CrossRef]
- Ramis, G.; Angeles Larrubia, M. An FT-IR study of the adsorption and oxidation of N-containing compounds over Fe2O3/Al2O3 SCR catalysts. J. Mol. Catal. A Chem. 2004, 215, 161–167. [Google Scholar] [CrossRef]
- Martínezarias, A.; Soria, J.; Conesa, J.C.; Seoane, X.L.; Arcoya, A.; Cataluna, R. NO reaction at surface oxygen vacancies generated in cerium oxide. J. Chem. Soc. Faraday Trans. 1995, 91, 1679–1687. [Google Scholar] [CrossRef]
- Kijlstra, W.S.; Brands, D.S.; Poels, E.K.; Bliek, A. Mechanism of the selective catalytic reduction of NO by NH3 over MnOx/Al2O3.1. Adsorption and desorption of the single reaction components. J. Catal. 1997, 171, 208–218. [Google Scholar] [CrossRef]
- Kijlstra, W.S.; Brands, D.S.; Smit, H.I.; Poels, E.K.; Bliek, A. Mechanism of the selective catalytic reduction of NO by NH3 over MnOx/Al2O3.2. Reactivity of adsorbed NH3 and NO complexes. J. Catal. 1997, 171, 219–230. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Knözinger, H. Species formed after NO adsorption and NO + O2 co-adsorption on TiO2: An FTIR spectroscopic study. Phys. Chem. Chem. Phys. 2000, 2, 2803–2806. [Google Scholar] [CrossRef]
- Kantcheva, M. Identification, stability, and reactivity of NOx species adsorbed on Titania-Supported manganese catalysts. J. Catal. 2001, 204, 479–494. [Google Scholar] [CrossRef]
- Qi, G.; Yang, R.T.; Chang, R. MnOx-CeO2, mixed oxides prepared by co-precipitation for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B Environ. 2004, 51, 93–106. [Google Scholar] [CrossRef]
- Long, R.Q.; Yang, R.T. Selective catalytic reduction of NO with Ammonia over Fe3+-Exchanged mordenite (Fe–MOR): Catalytic performance, characterization, and mechanistic Study. J. Catal. 2002, 207, 224–231. [Google Scholar] [CrossRef]
- Guan, B.; Lin, H.; Zhu, L.; Huang, Z. Selective catalytic reduction of NOx with NH3 over Mn, Ce substitution Ti0.9V0.1O2−δ nanocomposites catalysts prepared by self-propagating high-temperature synthesis method. J. Phys. Chem. C 2011, 115, 12850–12863. [Google Scholar] [CrossRef]
- Jung, S.M.; Grange, P. Investigation of the promotional effect of V2O5 on the SCR reaction and its mechanism on hybrid catalyst with V2O5 and TiO2-SO42− catalysts. Appl. Catal. B Environ. 2002, 36, 207–215. [Google Scholar] [CrossRef]
- Centeno, M.A.; Carrizosa, I.; Odriozola, J.A. In situ DRIFTS study of the SCR reaction of NO with NH3 in the presence of O2 over lanthanide doped V2O5/Al2O3 catalysts. Appl. Catal. B Environ. 1998, 19, 67–73. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. DRIFT study on Cerium–Tungsten/Titiania catalyst for selective catalytic reduction of NOx with NH3. Environ. Sci. Technol. 2010, 44, 9590–9596. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gas Composition | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| I | NH3 | NO | NH3 + NO | NH3 + NO + O2 |
| II | NH3 + O2 | NO | NH3 + NO + O2 | - |
| III | NO + O2 | NH3 + NO + O2 | NH3 + NO | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Yue, X.; Huang, T.; Shen, K.; Lu, B. In Situ DRIFTS Studies of NH3-SCR Mechanism over V2O5-CeO2/TiO2-ZrO2 Catalysts for Selective Catalytic Reduction of NOx. Materials 2018, 11, 1307. https://doi.org/10.3390/ma11081307
Zhang Y, Yue X, Huang T, Shen K, Lu B. In Situ DRIFTS Studies of NH3-SCR Mechanism over V2O5-CeO2/TiO2-ZrO2 Catalysts for Selective Catalytic Reduction of NOx. Materials. 2018; 11(8):1307. https://doi.org/10.3390/ma11081307
Chicago/Turabian StyleZhang, Yaping, Xiupeng Yue, Tianjiao Huang, Kai Shen, and Bin Lu. 2018. "In Situ DRIFTS Studies of NH3-SCR Mechanism over V2O5-CeO2/TiO2-ZrO2 Catalysts for Selective Catalytic Reduction of NOx" Materials 11, no. 8: 1307. https://doi.org/10.3390/ma11081307
APA StyleZhang, Y., Yue, X., Huang, T., Shen, K., & Lu, B. (2018). In Situ DRIFTS Studies of NH3-SCR Mechanism over V2O5-CeO2/TiO2-ZrO2 Catalysts for Selective Catalytic Reduction of NOx. Materials, 11(8), 1307. https://doi.org/10.3390/ma11081307