Formation and Thermal Behaviors of Ternary Silicon Oxycarbides derived from Silsesquioxane Derivatives

 ,
,
Abstract
:
1. Introduction

2. Experimental Section
2.1. Precursor Synthesis
2.2. Pyrolysis and Heat Treatment
2.3. Characterizations
3. Results and Discussion
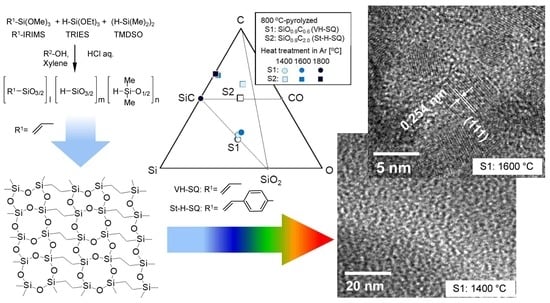
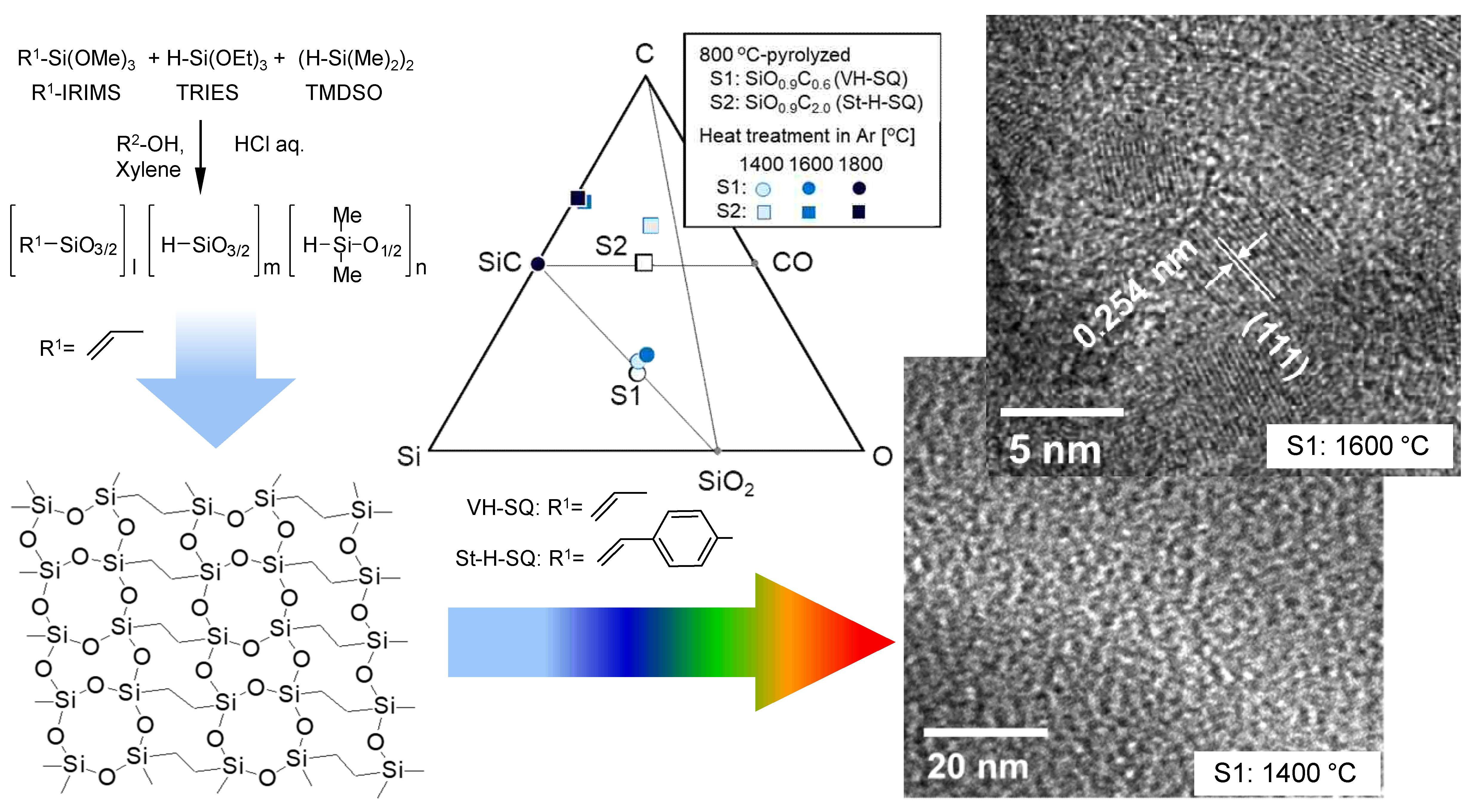
3.1. SQ Preceramic Polymers
3.2. Pyrolytic Behavior
3.3. Crystallization Behaviors of SQ-Derived Amorphous SiOC
4. Summary
- (1)
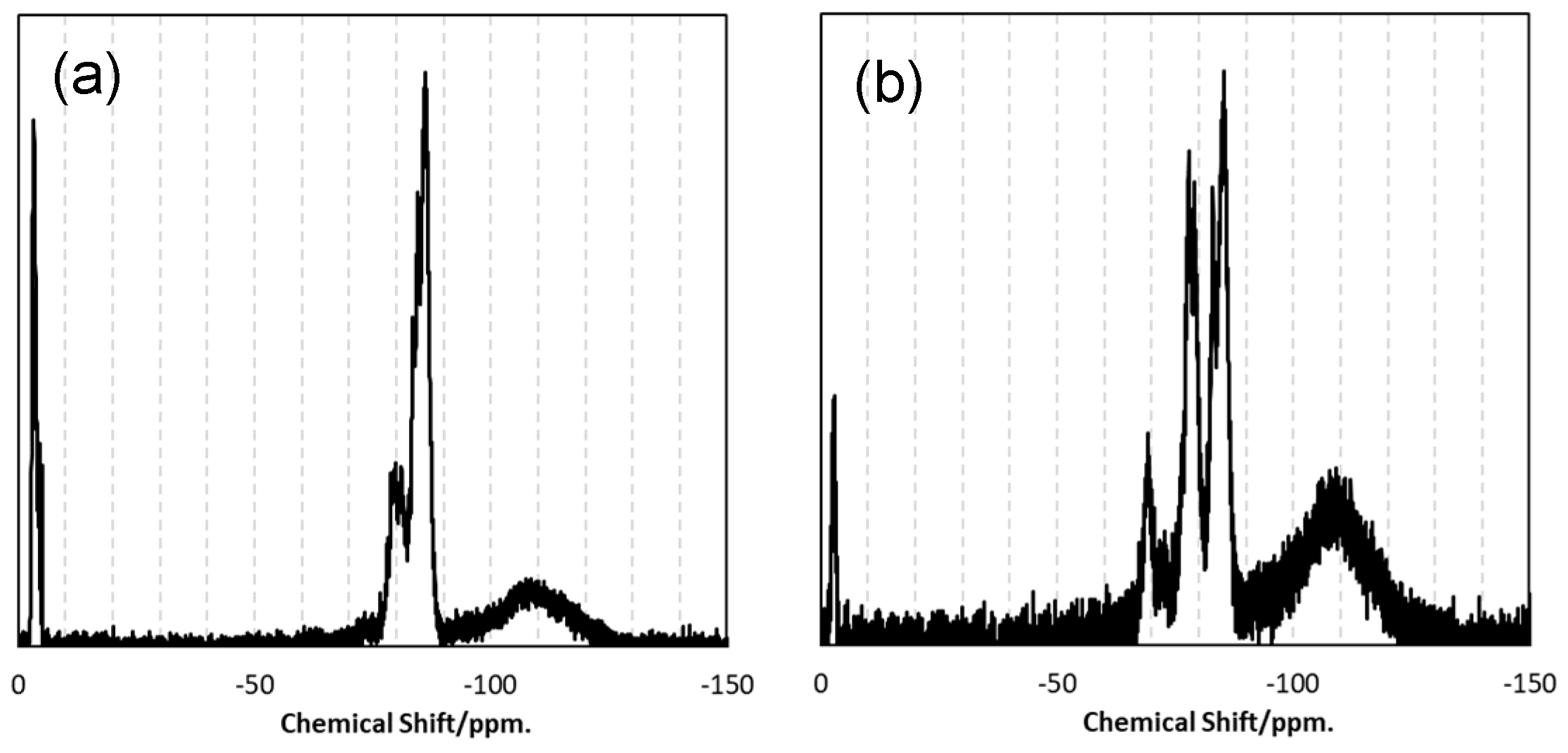
- ATR-IR and 29Si-NMR spectroscopic analyses revealed that the SQ derivatives VH-SQ (R = vinyl) and St-H-SQ (R = stylyl) were successfully synthesized through the conventional sol-gel route using R-Si(OMe)3, H-Si(OEt)3 and (H-Si(Me)2)2O as starting compounds.
- (2)
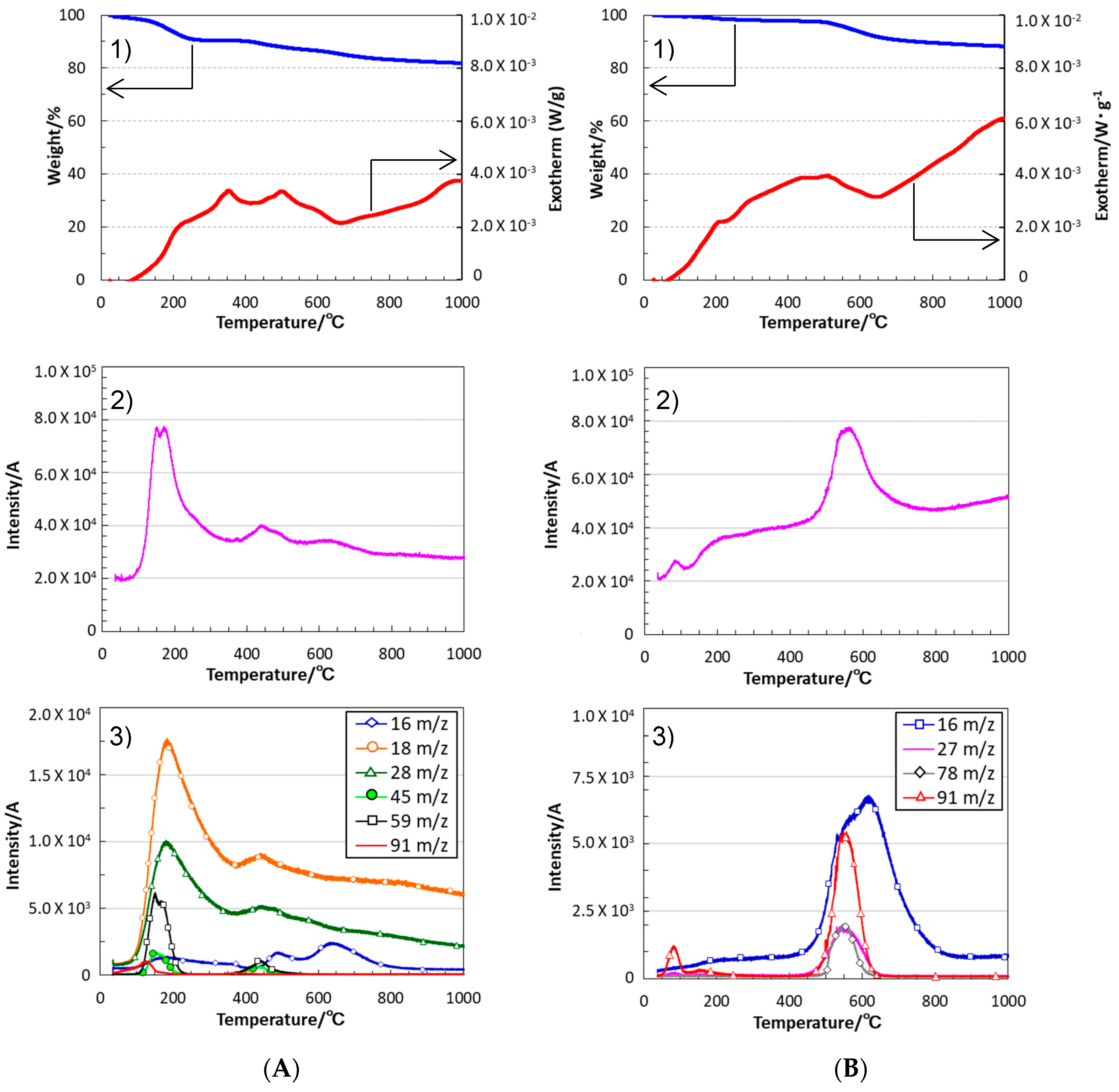
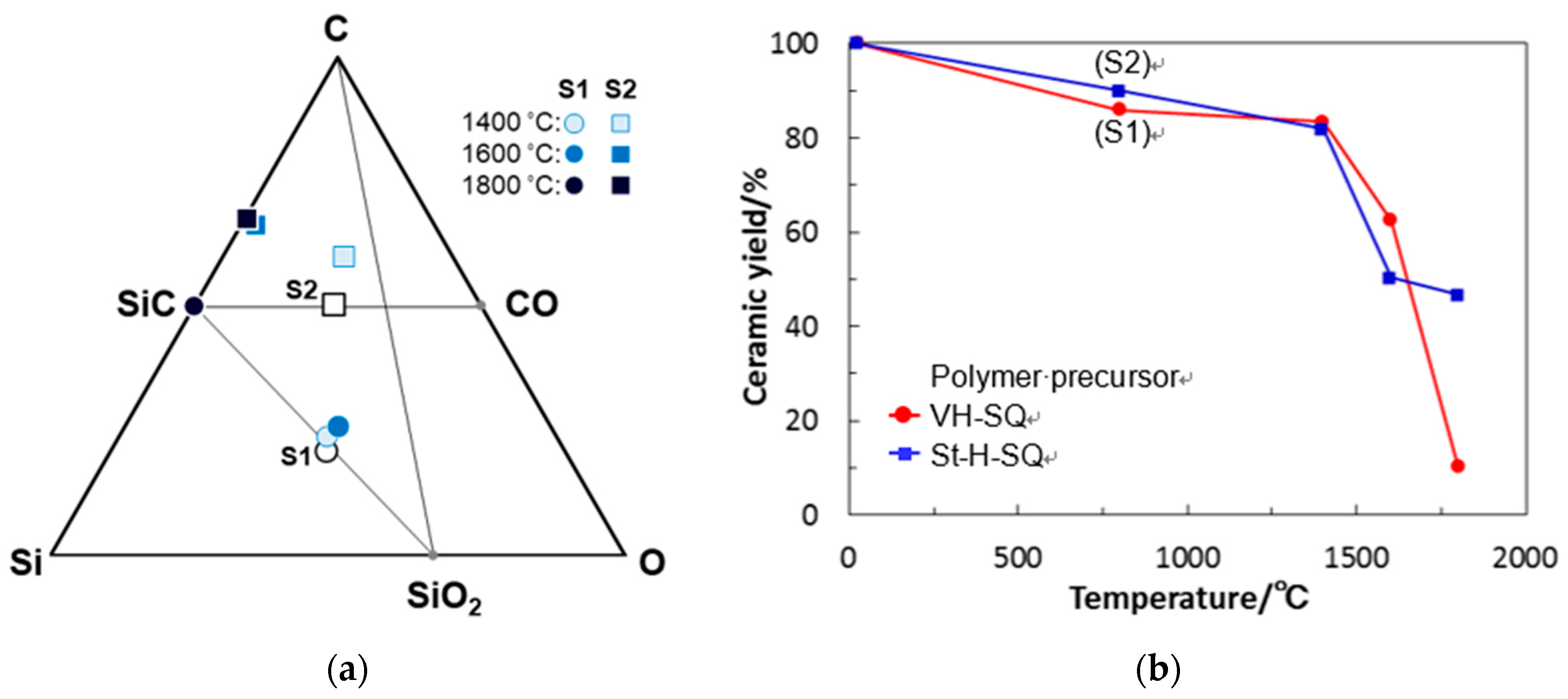
- The simultaneous TG-MS analyses showed that, under an inert atmosphere, thermally induced cross-linking via hydrosilylation and demethanation was efficiently achieved. The resulting ceramic yields after heating VH-SQ and St-H-SQ to 1000 °C were 82% and 88%, respectively.
- (3)
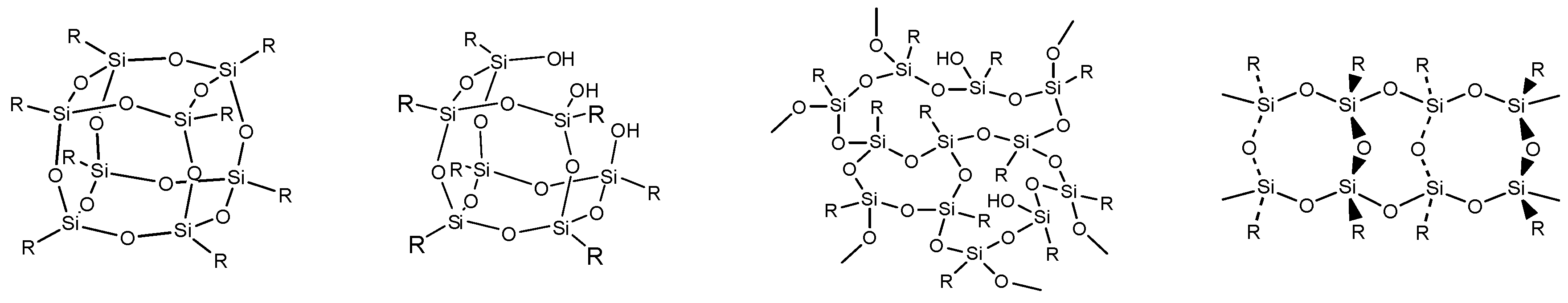
- Besides of the high ceramic yield, the in-situ cross-linking of VH-SQ led to suppressing the evolution of gaseous species that originated from hydrocarbon substituents up to 800 °C, leading to the formation of amorphous SiOC (labeled as S1) with a desired composition close to the stoichiometric SiO2(1−x)Cx (x = ca. 0.3).
- (4)
- By replacing the vinyl group with the styryl group, the carbon content in the amorphous SiOC successfully increased to afford SiO0.9C2.0H0.8 with excess carbon (labeled as S2).
- (5)
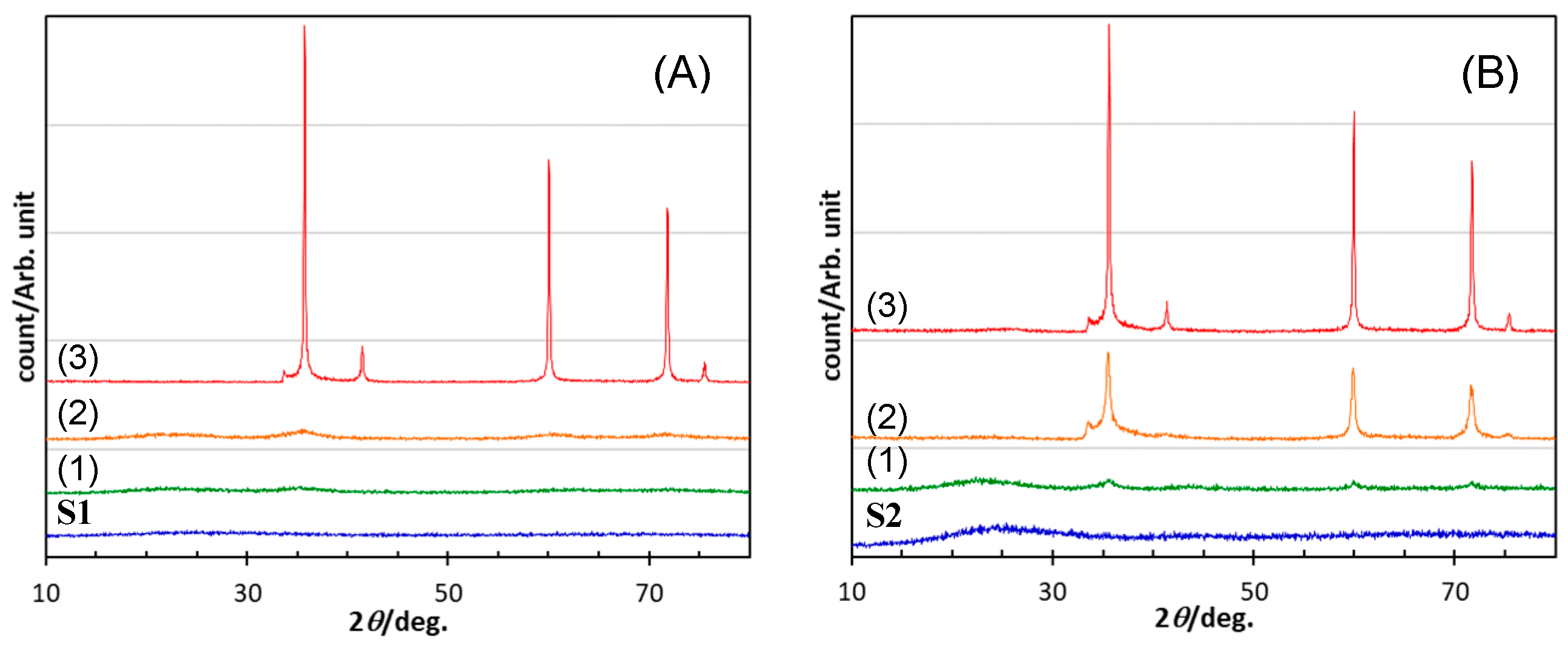
- Heat treatment up to 1800 °C in Ar of the amorphous SiOC materials revealed that, regardless of carbon content (in terms of stoichiometric S1 or S2 having excess carbon), the dominant pathway for β-SiC crystallization could be expressed by the following reactions: phase separation to amorphous/graphitic carbon and amorphous SiO2, followed by β-SiC crystallization via carbothermal reduction of SiO2.
- (6)
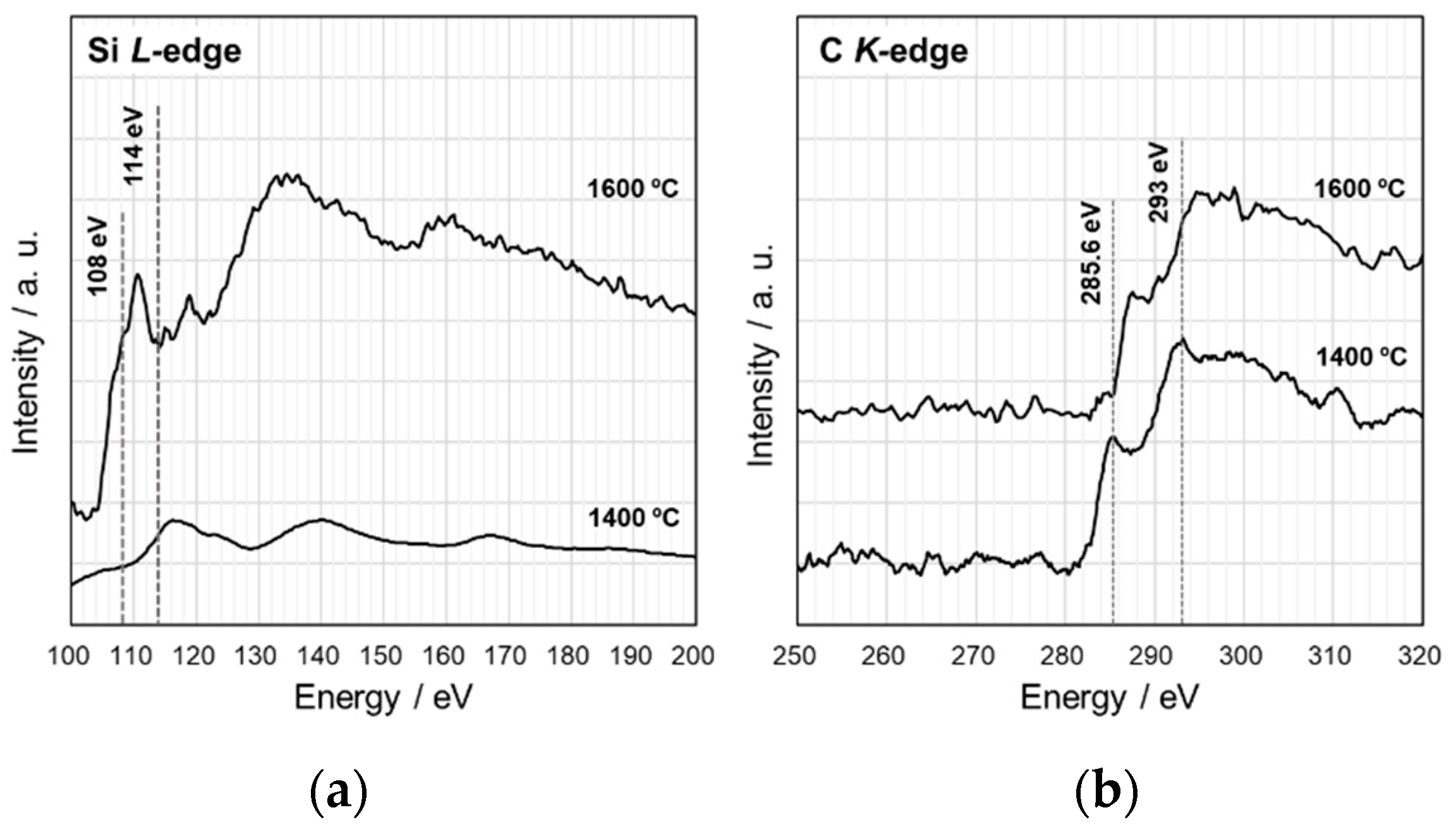
- The VH-SQ-derived S1 was found to hold an amorphous state up to 1400 °C. This enhanced thermal stability could be achieved by the phase separation associated with the local structure rearrangement from SiO2−xCx (x = 1, 2) to carbon-free SiO4 tetrahedral prior to the formation of SiC4 units essential for the nucleation and crystallization of β-SiC.
- (7)
- Further heat treatment above 1400 °C of the SQ-derived SiOC materials led to large weight loss and a remarkable decrease in oxygen content, which could be due to the thermodynamically favorable reaction of SiO2 with SiC to yield gaseous SiO and CO.
- (8)
- The VH-SQ was found to be a useful precursor for synthesizing amorphous SiOC with enhanced thermal stability. However, for the formation of β-SiC–SiO2 nanocomposite having a higher fraction of β-SiC nanocrystallites, it is important to facilitate the nucleation of SiC prior to phase separation to amorphous/graphitic carbon and amorphous SiO2. Currently, further study of the relationship between the chemical state of carbon and SiC nucleation within the amorphous SiOC, as well as heat treatment conditions to facilitate SiC nucleation, are in progress.
Author Contributions
Funding
Conflicts of Interest
References
- Díaz, U.; Corma, A. Organic-Inorganic Hybrid Materials: Multi-Functional Solids for Multi-Step Reaction Processes. Chem 2018, 24, 3944–3958. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.H.; Nagahara, L.A.; Thundat, T.; Mokarian-Tabari, P.; Furukawa, H.; Khosla, A. Review—Organic-Inorganic Hybrid Functional Materials: An Integrated Platform for Applied Technologies. J. Electrochem. Soc. 2018, 165, B3137–B3156. [Google Scholar] [CrossRef]
- Toagosei Co., Ltd. Organosilicon Compounds which have Oxetanyl Groups, and a Method for the Production and Curable Compositions of the Same. Patent No JP5454762, 26 March 2014. [Google Scholar]
- Kimoto, Y.; Fujita, T.; Furuta, N.; Kitamura, A.; Suzuki, H. Development of Space-Qualified Photocurable-Silsesquioxane-Coated Polyimide Films. J. Spacecr. Rocket. 2016, 53, 1028. [Google Scholar] [CrossRef]
- Toagosei Co., Ltd. Composition for Organic Semiconductor Insulating Films, and Organic Semiconductor Insulating Film. Patent No JP5704256, 22 April 2015. [Google Scholar]
- Toagosei Co., Ltd. Silicon-Containing Polymer Compound, Manufacturing Method of the same, Heat-Resistant Resin Composition, and Heat Resistant Coating. Patent No JP5601212, 8 October 2014. [Google Scholar]
- Blum, Y.D.; MacQueen, D.B.; Kleebe, H.-J. Synthesis and characterization of carbon-enriched silicon oxycarbides. J. Eur. Ceram. Soc. 2005, 25, 143–149. [Google Scholar] [CrossRef]
- Colombo, P.; Mera, G.; Riedel, R.; Soraru, G.D. Polymer-derived ceramics: 40 years of research and innovation in advanced ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Saha, A.; Raj, R. Crystallization Maps for SiCO Amorphous Ceramics. J. Am. Ceram. Soc. 2007, 90, 578–583. [Google Scholar] [CrossRef]
- Kleebe, H.-J.; Turquat, C.; Soraru, G.D. Phase Separation in an SiCO Glass Studied by Transmission Electron Microscopy and Electron Energy-Loss Spectroscopy. J. Am. Ceram. Soc. 2001, 84, 1073–1080. [Google Scholar] [CrossRef]
- Stabler, C.; Roth, F.; Narisawa, M.; Schliephake, D.; Heilmaier, M.; Lauterbach, S.; Kleebe, H.-J.; Riedel, R.; Ionescu, E. High-temperature creep behavior of a SiOC glass ceramic free of segregated carbon. J. Eur. Ceram. Soc. 2016, 35, 3747–3753. [Google Scholar] [CrossRef]
- Stabler, C.; Reitz, A.; Stein, P.; Albert, B.; Riedel, R.; Ionescu, E. Thermal Properties of SiOC Ceramics at Elevated Temperatures. Materials 2018, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A. Chapter 8, NMR Spectroscopy of Organosilicon Compounds. In The Chemistry of Organic Silicon Compounds Part 1; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1989; pp. 511–554. [Google Scholar]
- Pippel, E.; Lichtenberger, O.; Woltersdorf, J. Identification of silicon oxycarbide bonding in Si-C-O-glasses by EELS. J. Mater. Sci. Lett. 2000, 19, 2059–2060. [Google Scholar] [CrossRef]
- Wei, Q.; Pippel, E.; Woltersdorf, J.; Scheffler, M.; Greil, P. Interfacial SiC formation in polysiloxane-derived Si–O–C ceramics. Mater. Chem. Phys. 2002, 73, 281–289. [Google Scholar] [CrossRef]
- Savchenko, D.; Vasin, A.; Muto, S.; Kalabukhova, E.; Nazarov, A. EPR Study of Porous Si:C and SiO2:C Layers. Phys. Status Solidi B 2018, 255, 1700559. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef] [Green Version]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cancado, L.G.; Jorioa, A.; Saitoe, R. Studying Disorder in Graphite-Based Systems by Raman Spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, S.; Higashihira, M.; Maeda, K.; Tanaka, H. Raman Scattering Characterization of Polytype in Silicon Carbide Ceramics: Comparison with X-ray Diffraction. J. Am. Ceram. Soc. 2003, 86, 823–829. [Google Scholar] [CrossRef]
- Mutin, P.H. Role of Redistribution Reactions in the Polymer Route to Silicon–Carbon–Oxygen Ceramics. J. Am. Ceram. Soc. 2002, 85, 1185–1189. [Google Scholar] [CrossRef]
- Widgeon, S.J.; Sen, S.; Mera, G.; Ionescu, E.; Riedel, R.; Navrotsky, A. 29Si and 13C Solid-State NMR Spectroscopic Study of Nanometer-Scale Structure and Mass Fractal Characteristics of Amorphous Polymer Derived Silicon Oxycarbide Ceramics. Chem. Mater. 2010, 22, 6221–6228. [Google Scholar] [CrossRef]
- Ohyanagi, M.; Imai, T.; Toyofuku, N.; Nakagawa, D.; Munir, Z.A. Microscopic and Spectroscopic Characterization of Stacking-Sequence Disordered SiC. J. Am. Ceram. Soc. 2015, 98, 50–56. [Google Scholar] [CrossRef]
- Filsinger, D.H.; Bourrie, D.B. Silica to Silicon: Key Carbothermic Reactions and Kinetics. J. Am. Ceram. Soc. 1990, 73, 1726–1732. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition [wt%] | SiOC Composition Empirical Ratio SiO2(1−x)Cx + yCfree or ySifree | |||
|---|---|---|---|---|---|
| Si | O | C | H | ||
| As-synthesized VH-SQ | 45.9 | 34.1 | 15.7 | 4.26 | SiO1.3C0.8H2.6 (SiO1.3C0.35 + 0.45Cfree) |
| S1: 800 °C-pyrolyzed VH-SQ | 57.4 | 28.9 | 12.8 | 0.94 | SiO0.9C0.5H0.5 (SiO0.9C0.53 + 0.05Sifree) |
| S1: 1400 °C-heat treated | 55.4 | 29.5 | 15.1 | 0.08 | SiO0.9C0.6H0.0 SiO0.9C0.55 + 0.05Cfree |
| S1: 1600 °C-heat treated | 55.4 | 30.6 | 15.1 | 0.03 | SiO1.0C0.7H0.0 SiO0.9C0.50 + 0.2Cfree |
| S1: 1800 °C-heat treated | 55.4 | 0.24 | 29.7 | 0.02 | SiC1.0O0.0H0.0 SiC |
| As-synthesized St-H-SQ | 27.8 | 23.0 | 45.2 | 4.06 | SiO1.5C3.8H4.1 (SiO1.5C0.25 + 3.55Cfree) |
| S2: 800 °C-pyrolyzed St-H-SQ | 41.5 | 22.4 | 34.9 | 1.15 | SiO0.9C2.0H0.8 (SiO0.9C0.55 + 1.45Cfree) |
| S2: 1400 °C-heat treated | 33.4 | 21.2 | 45.4 | 0.13 | SiO1.1C3.2H0.1 SiO1.1C0.45 + 2.75Cfree |
| S2: 1600 °C-heat treated | 51.1 | 2.28 | 46.6 | 0.23 | SiO0.1C2.1H0.1 SiO1.0C0.95 + 1.15Cfree |
| S2: 1800 °C-heat treated | 52.5 | 0.28 | 47.2 | 0.04 | SiO0.0C2.1H0.0 SiC + 1.10Cfree |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwase, Y.; Fuchigami, T.; Horie, Y.; Daiko, Y.; Honda, S.; Iwamoto, Y. Formation and Thermal Behaviors of Ternary Silicon Oxycarbides derived from Silsesquioxane Derivatives. Materials 2019, 12, 1721. https://doi.org/10.3390/ma12101721
Iwase Y, Fuchigami T, Horie Y, Daiko Y, Honda S, Iwamoto Y. Formation and Thermal Behaviors of Ternary Silicon Oxycarbides derived from Silsesquioxane Derivatives. Materials. 2019; 12(10):1721. https://doi.org/10.3390/ma12101721
Chicago/Turabian StyleIwase, Yoshiaki, Teruaki Fuchigami, Yoji Horie, Yusuke Daiko, Sawao Honda, and Yuji Iwamoto. 2019. "Formation and Thermal Behaviors of Ternary Silicon Oxycarbides derived from Silsesquioxane Derivatives" Materials 12, no. 10: 1721. https://doi.org/10.3390/ma12101721
APA StyleIwase, Y., Fuchigami, T., Horie, Y., Daiko, Y., Honda, S., & Iwamoto, Y. (2019). Formation and Thermal Behaviors of Ternary Silicon Oxycarbides derived from Silsesquioxane Derivatives. Materials, 12(10), 1721. https://doi.org/10.3390/ma12101721