
Synthesis of High Crystallinity 1.13 nm Tobermorite and Xonotlite from Natural Rocks, Their Properties and Application for Heat-Resistant Products


Abstract
:1. Introduction
2. Materials and Methods
2.1. Characterization of the Raw Materials
2.2. Experimental Methods
2.2.1. Preparation of Samples
2.2.2. Instrumental Analysis
3. Results and Discussion
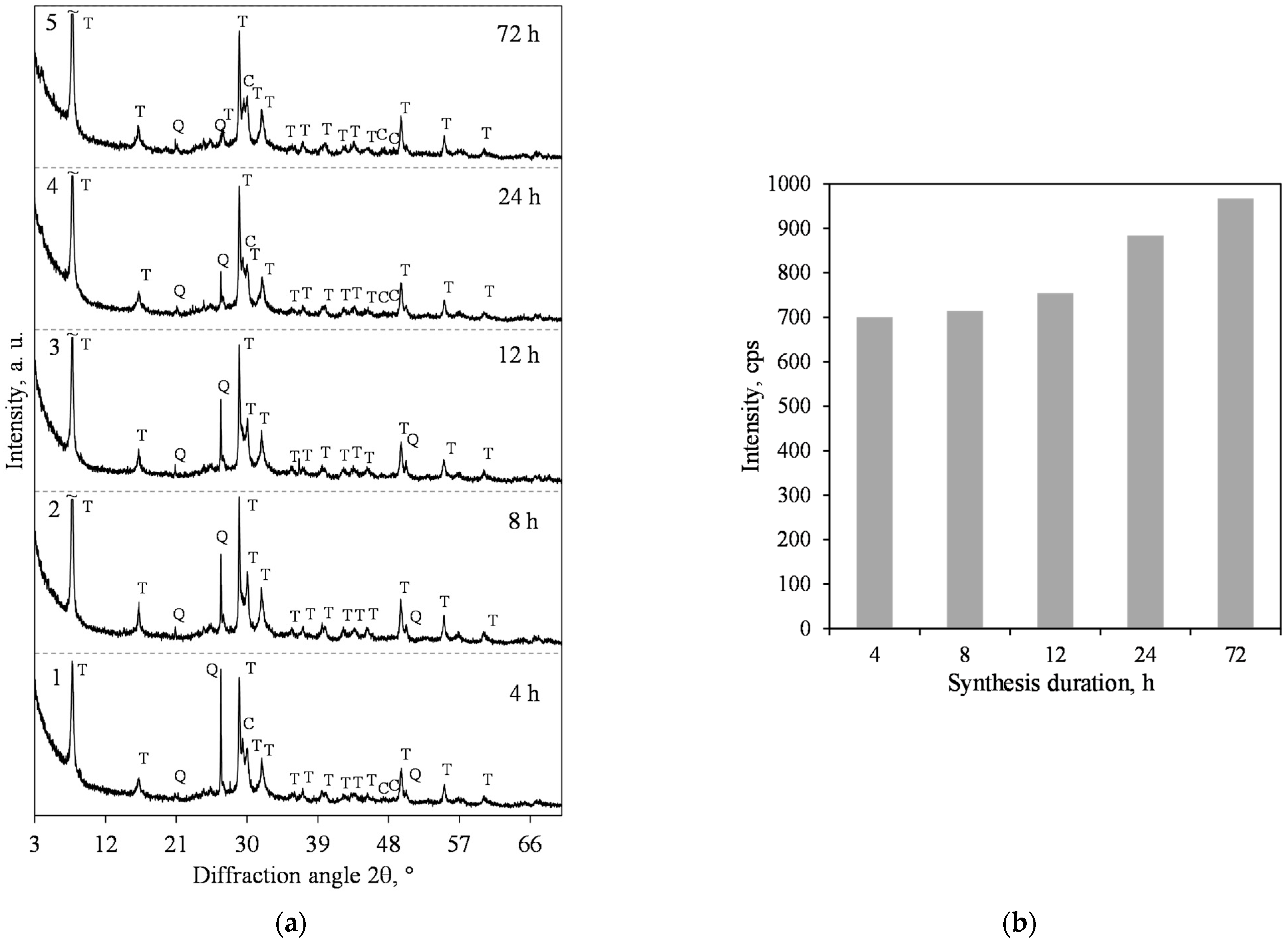
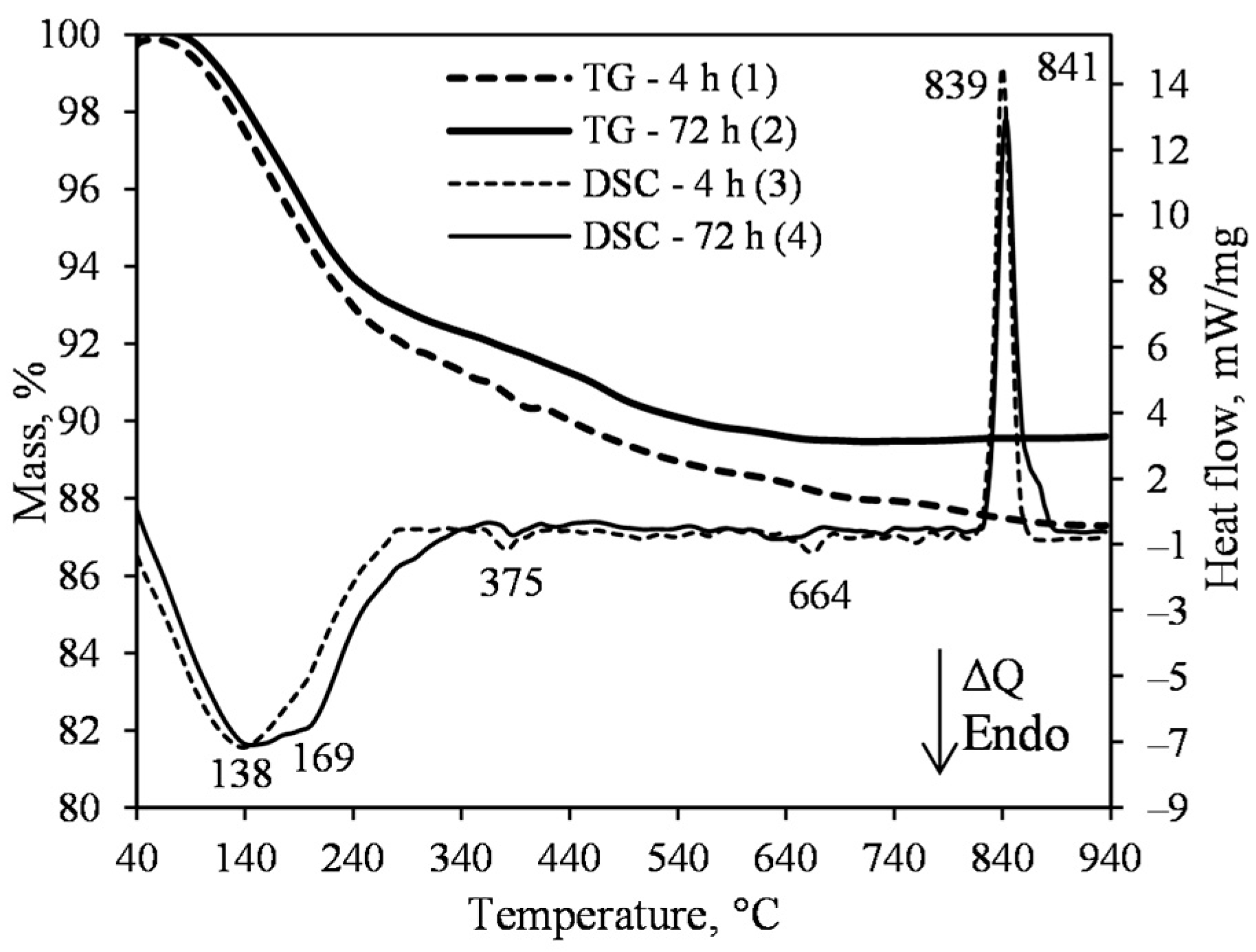
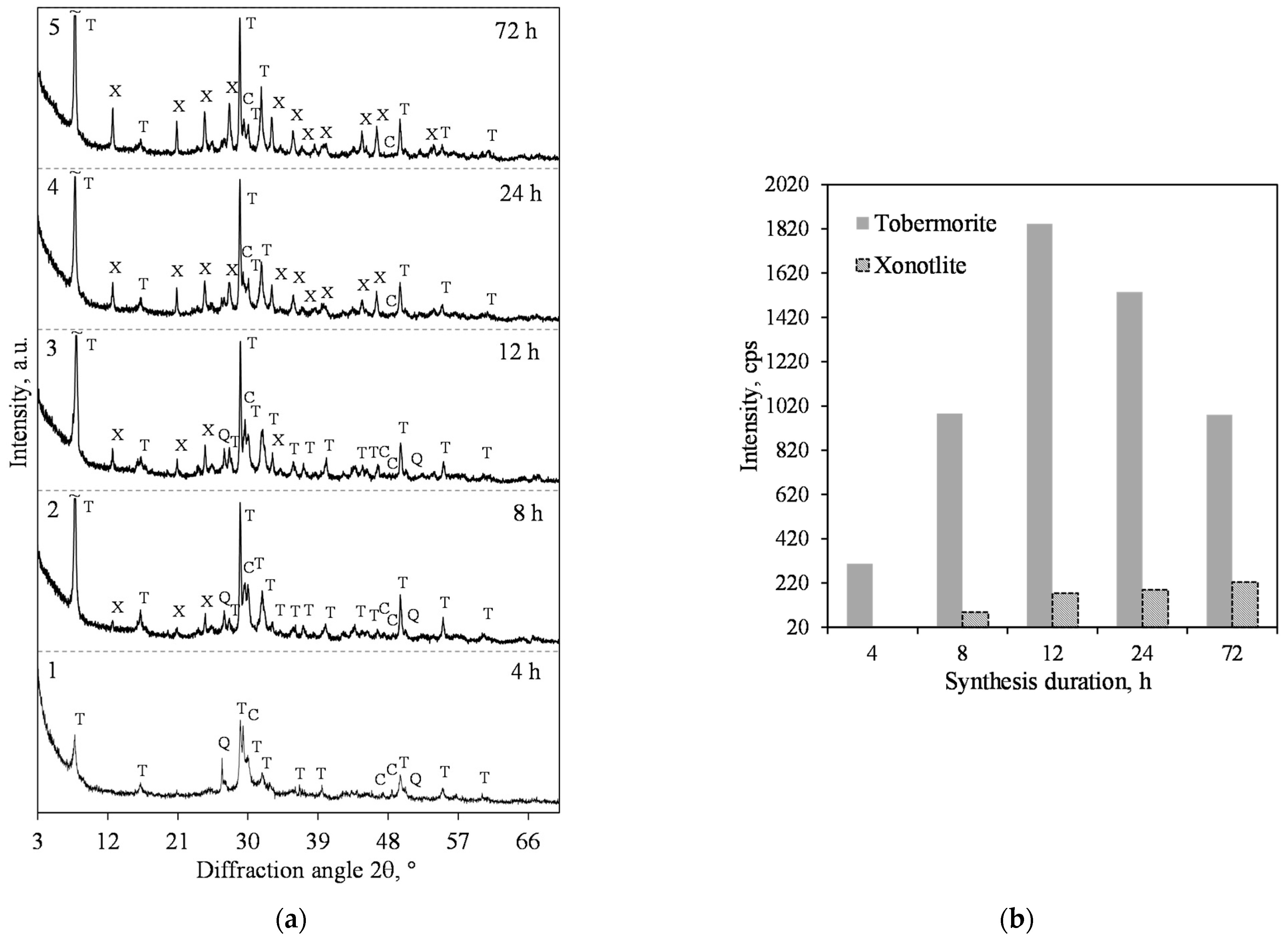
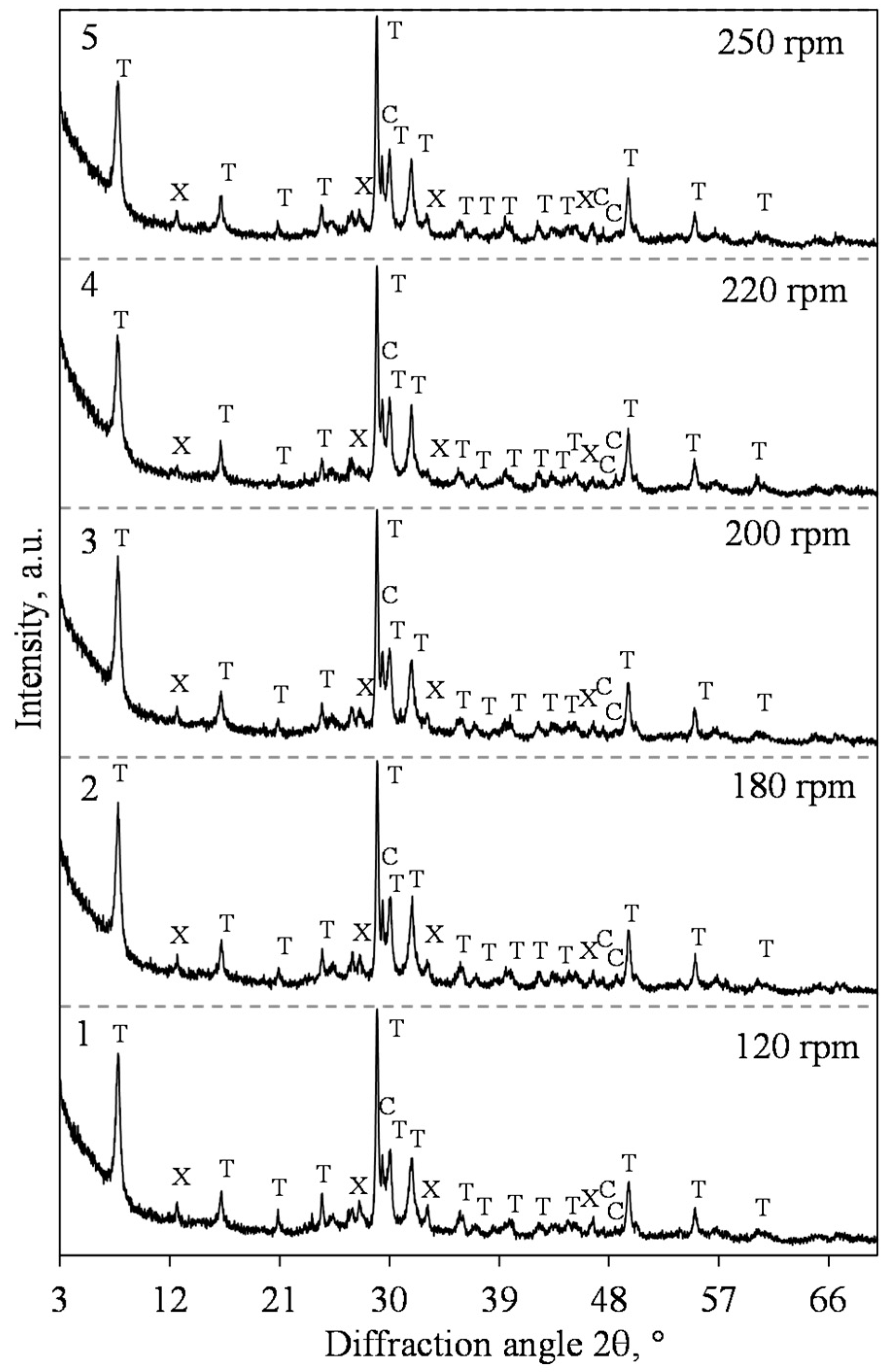
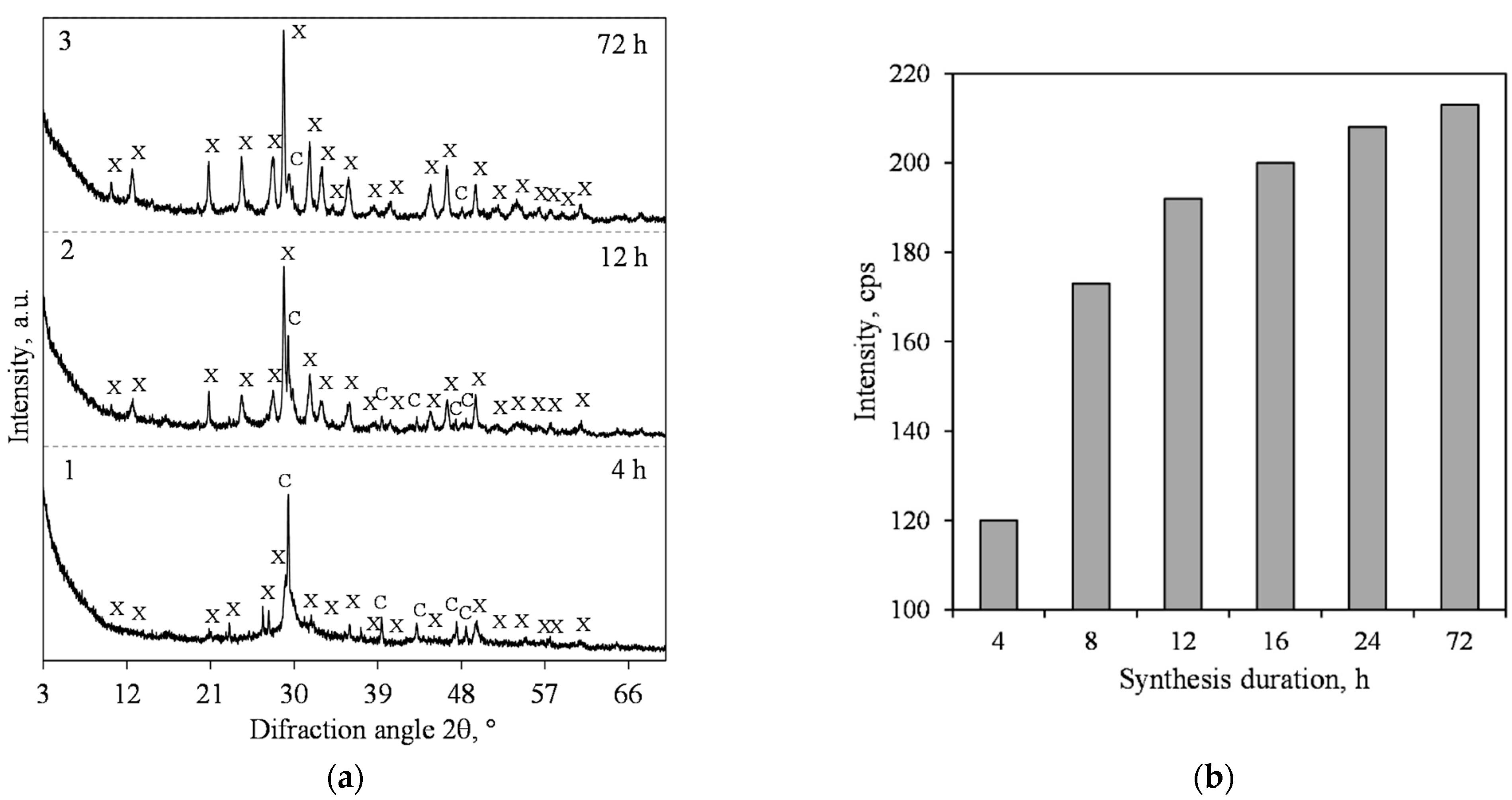
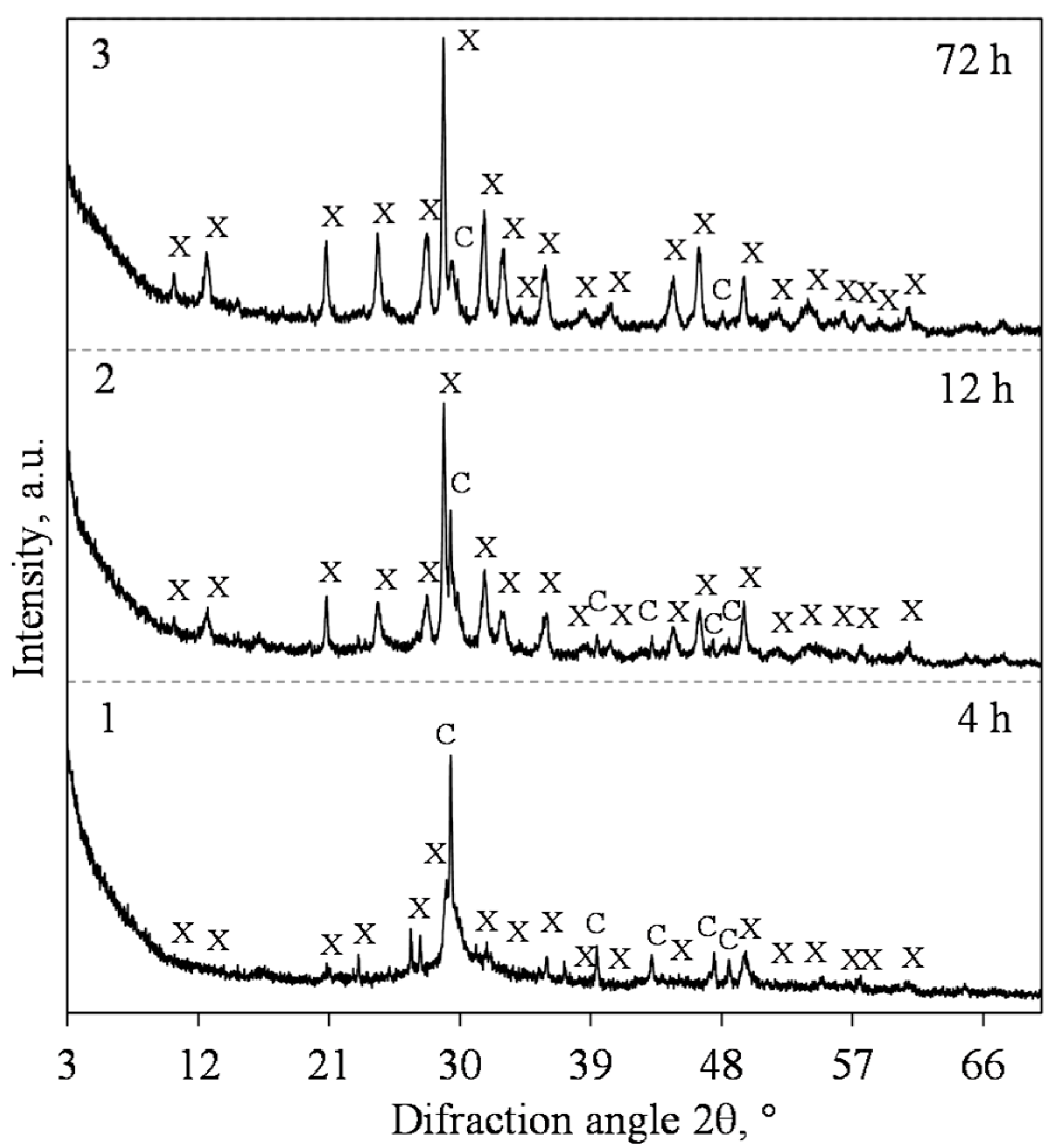
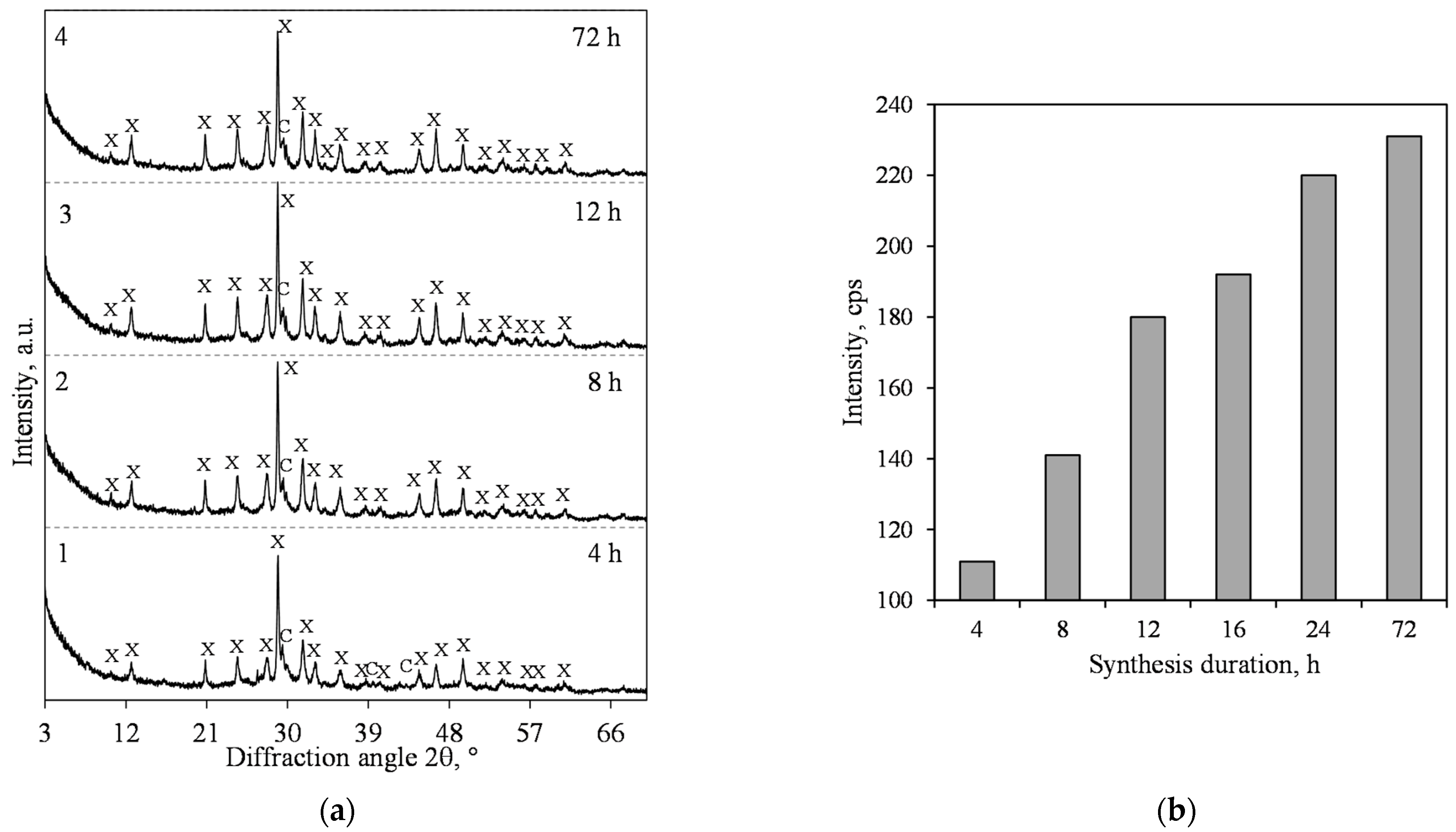
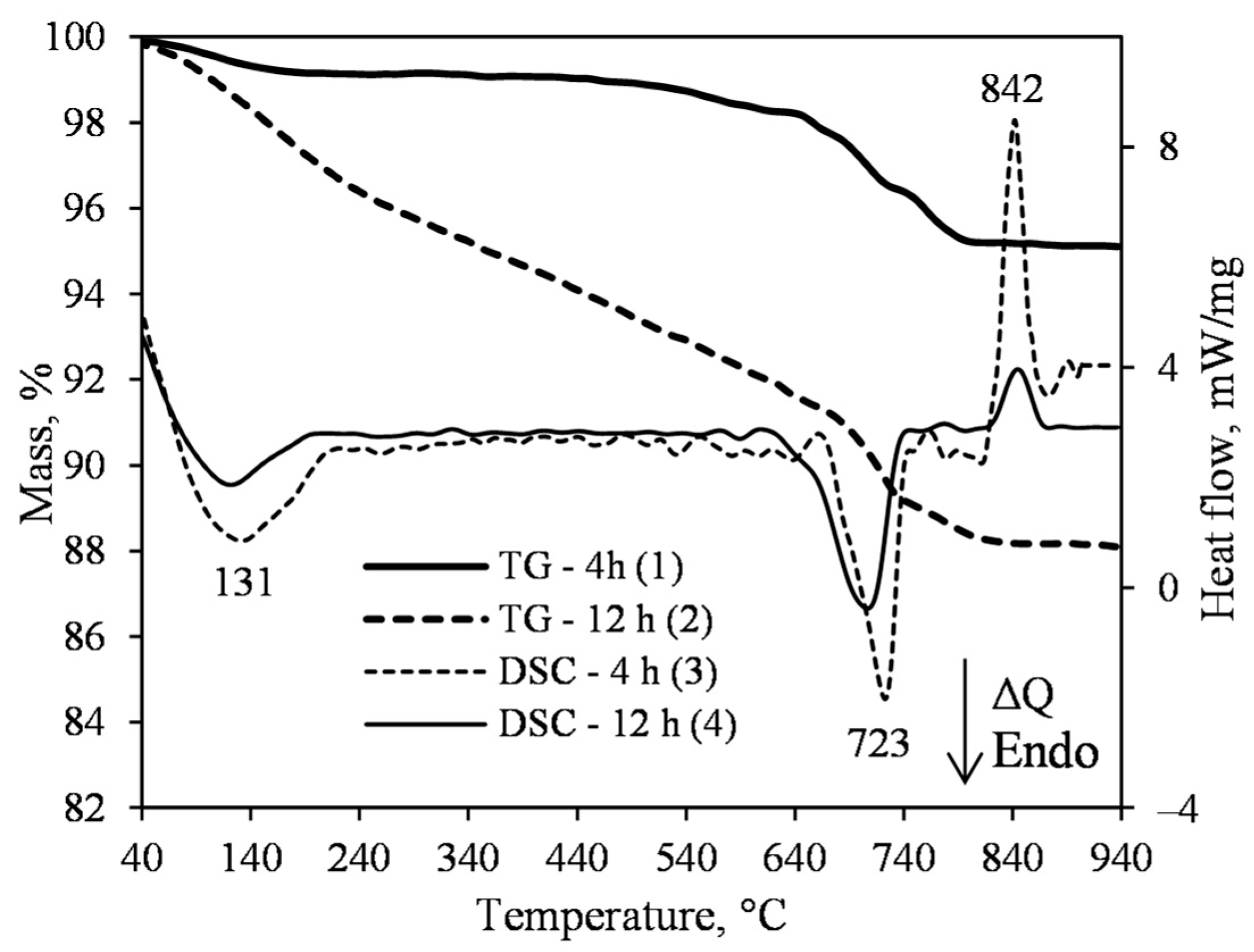
3.1. Hydrothermal Synthesis of 1.13 nm Tobermorite and Xonotlite
3.2. Evaluation of Suitability of Opoka for the Production of Heat-Resistant Thermal Insulating Materials from Calcium Silicate Hydrates
4. Conclusions
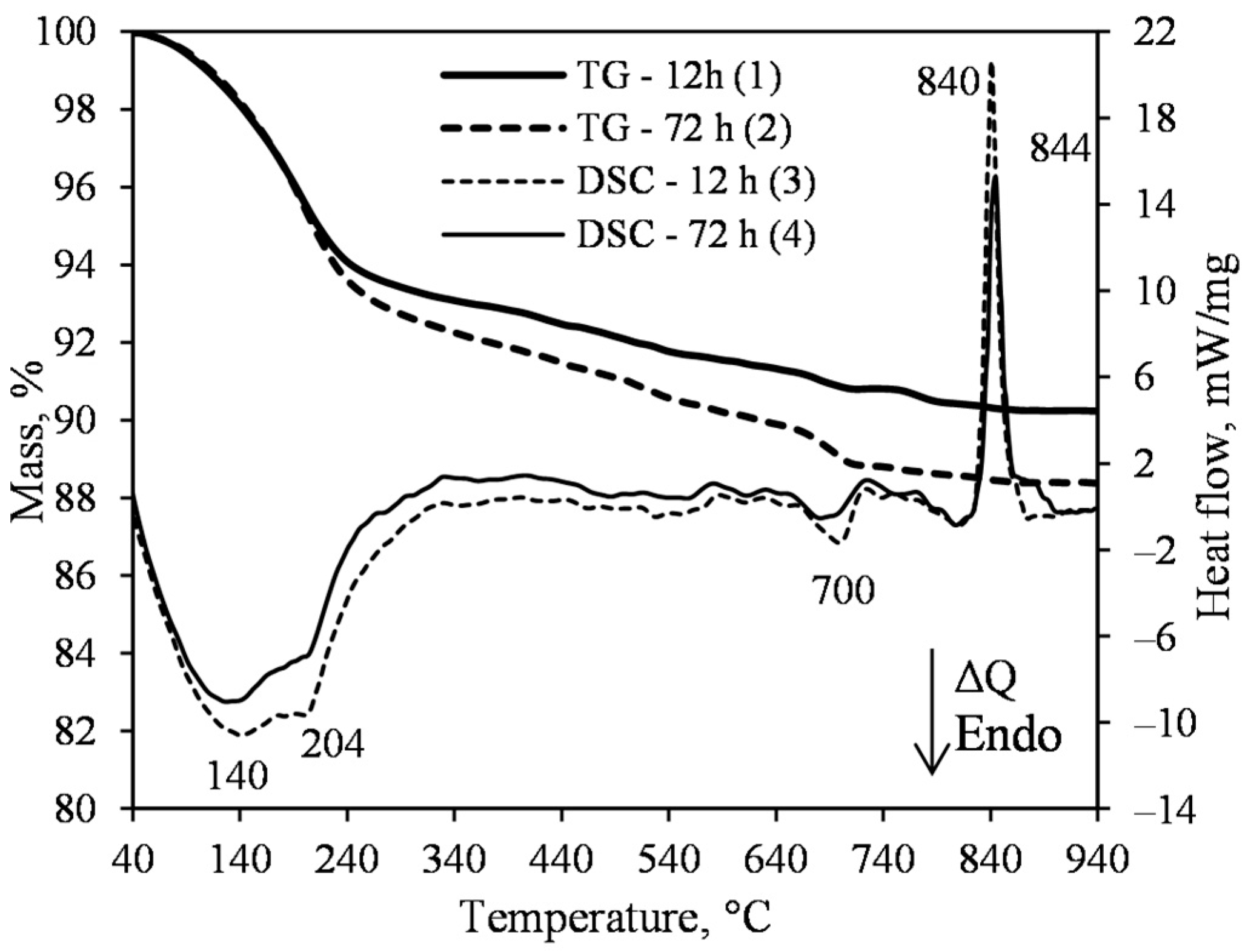
- The 1.13 nm tobermorite, with a high degree of crystallinity in the lime–calcined opoka mixture with a molar ratio of CaO/SiO2 = 0.83 at 200 °C, is formed rapidly at 4 h. However, even if the duration of the hydrothermal curing is extended to 72 h, a relatively high amount of semi-amorphous C–S–H (I) remains in the product. For this reason, tobermorite-based insulation products with good physical-mechanical properties (average density < 200 kg/m3, compressive strength ~0.9 MPa) but limited operating temperature (up to 700 °C) can be produced.
- The synthesis of xonotlite from industrial raw materials requires a very careful assessment of the amounts of impurities and their influence on the course of hydrothermal reactions. Impurities can react with CaO and alter the basicity of the reacting medium. In this case, the composition of the initial mixture needs to be adjusted.
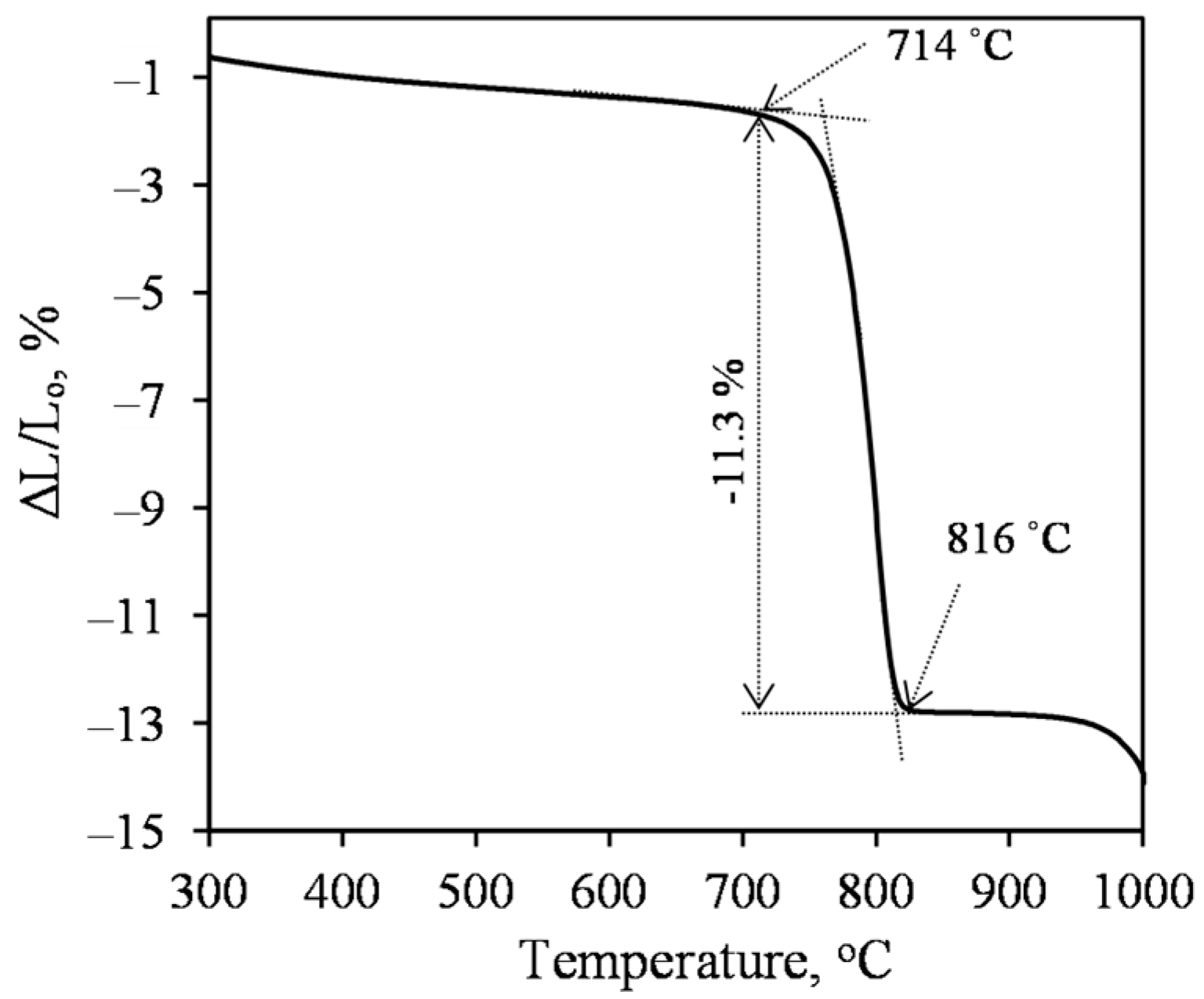
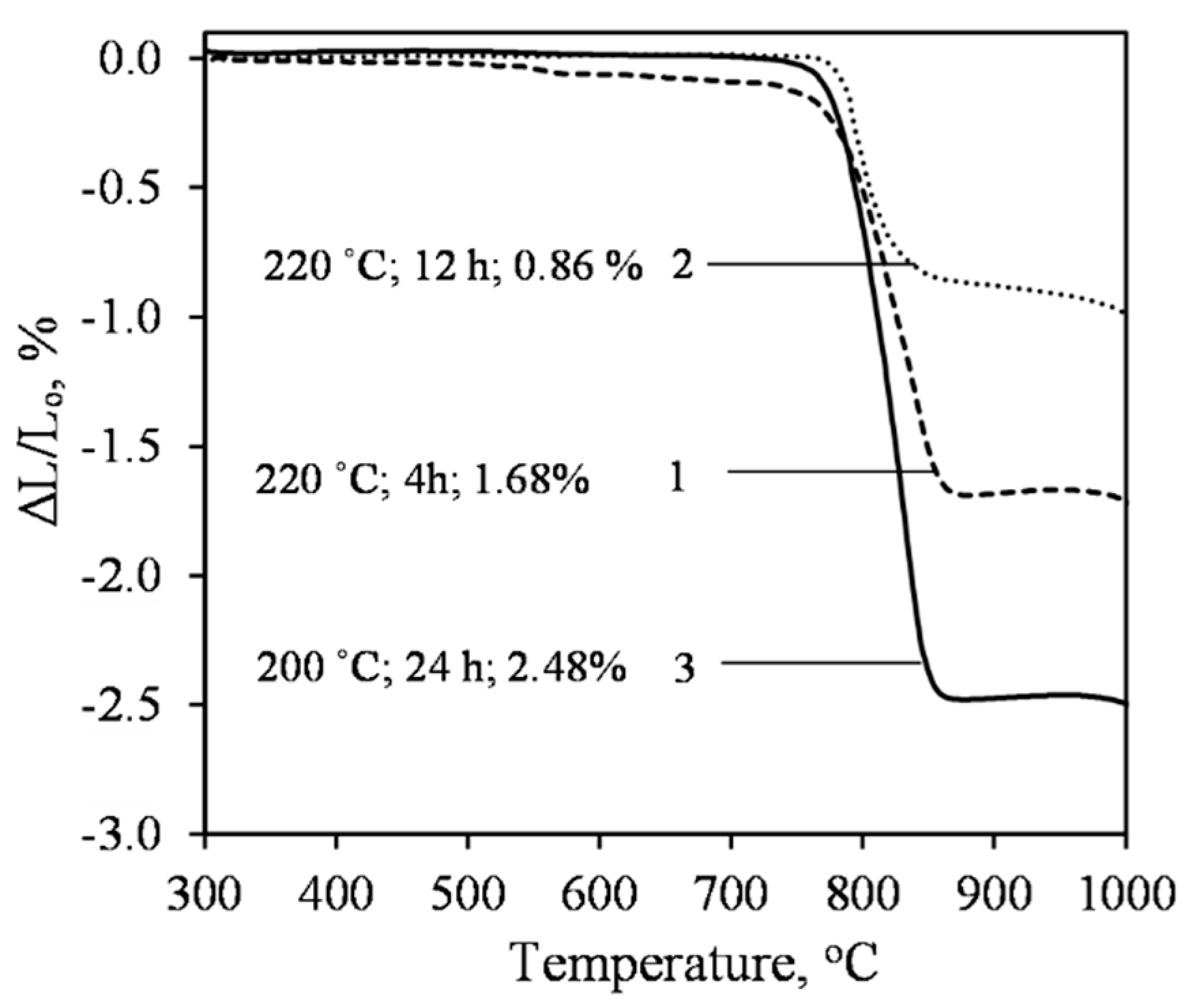
- Sufficiently pure xonotlite must be synthesized from calcined opoka to produce heat-resistant materials. Even a small amount of C–S–H(I) remaining in the product increases the linear shrinkage of the samples when burned to 1000 °C from 0.86 to 2% or more. Xonotlite precursors physically bind a large amount of water, resulting in very thick suspensions with a relative volume of about 20. It has been observed, however, that the thicker the suspension obtained during hydrothermal synthesis, the lower the density of the formed samples.
- High-quality heat-resistant (up to at least 1000 °C) xonotlite-based products can be made from lime–calcined opoka mixtures. Our recommended parameters are as follows: a molar ratio of the initial mixture of CaO/SiO2 = 1.2; a water/solid ratio of suspension W/S = 20.0; the duration of hydrothermal synthesis to be set at 220 °C for 8 h and the duration of autoclaving to be set at 220 °C for 4 h. The relative volume of the suspension is ~20, the average density of the obtained samples is ~180 kg/m3, and the compressive strengths exceed 1.5 MPa.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coleman, N.J.; Brassington, D.S.; Raza, A.; Mendham, A.P. Sorption of Co2+ and Sr2+ by waste-derived 11 Å tobermorite. Waste Manag. 2006, 26, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Komarneni, S.; Roy, D.M. Tobermorites: A New Family of Cation Exchangers. Science 1983, 221, 647–648. [Google Scholar] [CrossRef] [PubMed]
- Komarneni, S.; Roy, D.M. New tobermorite cation exchangers. J. Mater. Sci. 1985, 20, 2930–2936. [Google Scholar] [CrossRef]
- Paradiso, P.; Santos, R.L.; Horta, R.B.; Lopes, J.N.C.; Ferreira, P.J.; Colaço, R. Formation of nanocrystalline tobermorite in calcium silicate binders with low C/S ratio. Acta Mater. 2018, 152, 7–15. [Google Scholar] [CrossRef]
- Grangeon, S.; Claret, F.; Roosz, C.; Sato, T.; Gaboreau, S.; Linard, Y. Structure of nanocrystalline calcium silicate hydrates: Insights from X-ray diffraction, synchrotron X-ray absorption and nuclear magnetic resonance. J. Appl. Cryst. 2016, 49, 771–783. [Google Scholar] [CrossRef]
- Frost, R.L.; Mahendran, M.; Poologanathan, K.; Xi, Y. Raman spectroscopic study of the mineral xonotlite Ca6Si6O17(OH)2—A component of plaster boards. Mater. Res. Bull. 2012, 47, 3644–3649. [Google Scholar] [CrossRef] [Green Version]
- Luke, K.; Taylor, H.F.W.; Kalousek, G.L. Some factors affecting formation of truscottite and xonotlite at 300–350 °C. Cem. Concr. Res. 1981, 11, 197–203. [Google Scholar] [CrossRef]
- Liu, F.; Cao, J.X.; Zhu, B. Effect of anion impurity on preparing xonotlite whiskers via hydrothermal synthesis. Adv. Mat. Res. 2011, 148–149, 1755–1758. [Google Scholar] [CrossRef]
- Pugovkina, Y.; Kutugin, V.; Ostroumova, A.; Rymanova, I. High temperature and heat insulated calcium silicate materials. Key Eng. Mater. 2016, 683, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Akbayrak, S.; Özkar, S. Inverse relation between the catalytic activity and catalyst concentration for the ruthenium(0) nanoparticles supported on xonotlite nanowire in hydrogen generation from the hydrolysis of sodium borohydride. J. Mol. Catal. A Chem. 2016, 424, 254–260. [Google Scholar] [CrossRef]
- Black, L.; Garbev, K.; Stemmermann, P.; Hallam, K.R.; Allen, G.C. Characterisation of crystalline C-S-H phases by X-ray photoelectron spectroscopy. Cem. Concr. Res. 2003, 33, 899–911. [Google Scholar] [CrossRef]
- Ding, J.; Tang, Z.; Ma, S.; Wang, Y.; Zheng, S.; Zhang, Y.; Shen, S.; Xie, Z. A novel process for synthesis of tobermorite fiber from high-alumina fly ash. Cem. Concr. Compos. 2016, 65, 11–18. [Google Scholar] [CrossRef]
- Zou, J.; Guo, C.; Jiang, Y.; Wei, C.; Li, F. Structure, morphology and mechanism research on synthesizing xonotlite fiber from acid-extracting residues of coal fly ash and carbide slag. Mater. Chem. Phys. 2016, 172, 121–128. [Google Scholar] [CrossRef]
- Guo, X.; Meng, F.; Shi, H. Microstructure and characterization of hydrothermal synthesis of Al-substituted tobermorite. Constr. Build. Mater. 2017, 133, 253–260. [Google Scholar] [CrossRef]
- Grangeon, S.; Claret, F.; Lerouge, C.; Warmont, F.; Sato, T.; Anraku, S.; Numako, C.; Linard, Y.; Lanson, B. On the nature of structural disorder in calcium silicate hydrates with a calcium/silicon ratio similar to tobermorite. Cem. Concr. Res. 2013, 52, 31–37. [Google Scholar] [CrossRef]
- Hartmann, A.; Khakhutov, M.; Buhl, J.-C. Hydrothermal synthesis of CSH-phases (tobermorite) under influence of Ca-formate. Mater. Res. Bull. 2014, 51, 389–396. [Google Scholar] [CrossRef]
- Kikuma, J.; Tsunashima, M.; Ishikawa, T.; Matsuno, S.-Y.; Ogawa, A.; Matsui, K.; Sato, M. In situ Time-Resolved X-ray diffraction of tobermorite formation process under autoclave condition. J. Am. Ceram. Soc. 2010, 93, 2667–2674. [Google Scholar] [CrossRef]
- Eℓ-Hemaly, S.; Mitsuda, T.; Taylor, H. Synthesis of normal and anomalous tobermorites. Cem. Concr. Res. 1977, 7, 429–438. [Google Scholar] [CrossRef]
- Chan, C.; Mitsuda, T. Formation of 11 Å tobermorite from mixtures of lime and colloidal silica with quartz. Cem. Concr. Res. 1978, 8, 135–138. [Google Scholar] [CrossRef]
- Galvánková, L.; Másilko, J.; Solný, T.; Štěpánková, E. Tobermorite synthesis under hydrothermal conditions. Procedia Eng. 2016, 151, 100–107. [Google Scholar] [CrossRef]
- Ríos, C.A.; Williams, C.D.; Fullen, M.A. Hydrothermal synthesis of hydrogarnet and tobermorite at 175 °C from kaolinite and metakaolinite in the CaO–Al2O3–SiO2–H2O system: A comparative study. Appl. Clay Sci. 2009, 43, 228–237. [Google Scholar] [CrossRef]
- Liu, S.K.; Han, C.; Liu, J.M.; Li, H. Hydrothermal decomposition of potassium feldspar under alkaline conditions. RSC Adv. 2015, 5, 93301–93309. [Google Scholar] [CrossRef]
- Youssef, H.; Ibrahim, D.; Komarneni, S.; Mackenzie, K.J.D. Synthesis of 11 Å Al-substituted tobermorite from trachyte rock by hydrothermal treatment. Ceram. Int. 2010, 36, 203–209. [Google Scholar] [CrossRef]
- Maeshima, T.; Noma, H.; Sakiyama, M.; Mitsuda, T. Natural 1.1 and 1.4 nm tobermorites from Fuka, Okayama, Japan: Chemical analysis, cell dimensions, 29Si NMR and thermal behavior. Cem. Concr. Res. 2003, 33, 1515–1523. [Google Scholar] [CrossRef]
- Smalakys, G.; Siauciunas, R. The synthesis of 1.13 nm tobermorite from carbonated opoka. J. Therm. Anal. Calorim. 2018, 134, 493–502. [Google Scholar] [CrossRef]
- Panda, S.; Biswal, A.; Mishra, S.; Panda, P.K.; Pradhan, N.; Mohapatra, U.; Sukla, L.B.; Mishra, B.K.; Akcil, A. Reductive dissolution by waste newspaper for enhanced meso-acidophilic bioleaching of copper from low grade chalcopyrite: A new concept of biohydrometallurgy. Hydrometallurgy 2015, 153, 98–105. [Google Scholar] [CrossRef]
- Jiménez, I.; Pérez, G.; Guerrero, A.; Ruiz, B. Mineral phases synthesized by hydrothermal treatment from biomass ashes. Int. J. Miner. Process. 2017, 158, 8–12. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Nishimoto, S.; Kameshima, Y.; Miyake, M. Hydrothermal preparation of tobermorite from blast furnace slag for Cs+ and Sr2+ sorption. J. Hazard. Mater. 2014, 266, 174–181. [Google Scholar] [CrossRef]
- Wang, S.; Peng, X.; Tang, L.; Zeng, L.; Lan, C. Influence of inorganic admixtures on the 11 Å-tobermorite formation prepared from steel slags: XRD and FTIR analysis. Constr. Build. Mater. 2014, 60, 42–47. [Google Scholar] [CrossRef]
- Hartmann, A.; Schulenberg, D.; Buhl, J.-C. Synthesis and structural characterization of CSH-phases in the range of C/S= 0.41–1.66 at temperatures of the tobermorite xonotlite crossover. J. Mater. Sci. Chem. Eng. 2015, 3, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Chen, S.; Lin, Q.; Wang, X.D.; Cao, J.X. Study on hydrothermal synthesis dynamics of nanoscale xonotlite fibers. IOP Conf. Ser. Mater. Sci. Eng. 2018, 292, 012050. [Google Scholar] [CrossRef]
- Garbev, K.; Black, L.; Beuchle, G.; Stemmermann, P. Inorganic polymers in cement based materials. Wasser-Und Geotechnol. 2002, 1, 19–30. [Google Scholar]
- Shaw, S.; Clark, S.M.; Henderson, C.M.B. Hydrothermal formation of the calcium silicate hydrates, tobermorite (Ca5Si6O16(OH)2·4H2O) and xonotlite (Ca6Si6O17(OH)2): An in situ synchrotron study. Chem. Geol. 2000, 167, 129–140. [Google Scholar] [CrossRef]
- Kalousek, G.L.; Mitsuda, T.; Taylor, H.F.W. Xonotlite: Cell parameters, thermogravimetry and analytical electron microscopy. Cem. Concr. Res. 1977, 7, 305–312. [Google Scholar] [CrossRef]
- Liu, F.; Chen, S.; Lin, Q.; Wang, X.-D.; Cao, J.-X. Effect of anions from calcium sources on the synthesis of nano-sized xonotlite fibers. Optoelectron. Lett. 2017, 13, 81–83. [Google Scholar] [CrossRef]
- Lin, J.; Li, G.; Liu, W.; Qiu, R.; Wei, H.; Zong, K.; Cai, X. A review of recent progress on the silica aerogel monoliths: Synthesis, reinforcement, and applications. J Mater. Sci. 2021, 56, 10812–10833. [Google Scholar] [CrossRef]
- Liu, F.; Zeng, L.; Cao, J.; Li, J. Preparation of ultra-light xonotlite thermal insulation material using carbide slag. J. Wuhan Univ. Technol. 2010, 25, 295–297. [Google Scholar] [CrossRef]
- Standard EN 16977; Thermal Insulation Products for Buildings—Factory Made Calcium Silicate (CS) Products—Specification. Technical Committee CEN/TC 88 “Thermal INSULATING Materials and Products”: Brussels, Belgium, 2020; pp. 1–34.
- Biagoni, C.; Bonaccorsi, E.; Lezzerini, M.; Merlino, S. Thermal behaviour of Al-rich tobermorite. Eur. J. Miner. 2016, 28, 23–32. [Google Scholar] [CrossRef]
- Siauciunas, R.; Mikaliunaite, J.; Urbonas, L.; Baltakys, K. Tribochemical and thermal activation of α-C2S hydrate as precursor for cementitious binders. J. Therm. Anal. Calorim. 2014, 118, 817–823. [Google Scholar] [CrossRef]
- Baltakys, K.; Siauciunas, R. The influence of γ-Al2O3 and Na2O on the formation of gyrolite in the stirring suspension. J. Mater. Sci. 2006, 41, 4799–4805. [Google Scholar] [CrossRef]
- Smalakys, G.; Siauciunas, R. Peculiarities of xonotlite synthesis from the raw materials with different SiO2 activities. J. Therm. Anal. Calorim. 2020, 142, 1671–1679. [Google Scholar] [CrossRef]
- Cao, J.; Liu, F.; Lin, Q.; Zhang, Y. Hydrothermal synthesis of xonotlite from carbide slag. Prog. Nat. Sci. 2008, 18, 1147–1153. [Google Scholar] [CrossRef]
- Cao, J.X.; Zhang, Y.; Zeng, L.K. Preparation of nanoporous super thermal insulation material compounded with xonotlite-SiO2-aerogel and characterization of the pore structure. Key Eng. Mater. 2007, 336–338, 1505–1508. [Google Scholar] [CrossRef]
- Taylor, H.F.W. Tobermorite, jennite, and cement gel. Z. Krist. -Cryst. Mater. 1992, 202, 41–50. [Google Scholar] [CrossRef]
- Shaw, S.; Henderson, C.M.B.; Komanschek, B.U. Dehydration/recrystallization mechanisms, energetics, and kinetics of hydrated calcium silicate minerals: An in situ TGA/DSC and synchrotron radiation SAXS/WAXS study. Chem. Geol. 2000, 167, 141–159. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System No. | Mixture with Molar Ratio CaO/SiO2 | The Temperature of the Isothermal Curing, °C | Duration of Isothermal Curing, h | The Suspension Stirred Rate, rpm |
|---|---|---|---|---|
| 1 | 0.83 | 200 | 4; 8; 12; 24; 72 | 50 |
| 2 | 1 | 200 | 4; 8; 12; 24; 72 | 50 |
| 3 | 1 | 200 | 12 | 120; 180; 200; 220; 250;300 |
| 4 | 1.0 | 220 | 4; 8; 12; 16; 24; 72 | 300 |
| 5 | 1.2 | 200 | 4; 12; 72 | 300 |
| 6 | 1.2 | 220 | 4; 8; 12; 16; 24; 72 | 300 |
| Stirring rate, rpm | 50 | 120 | 180 | 200 | 220 | 250 | 300 |
| Relative volume, cm3/g | 6 | 7 | 7.5 | 10 | 11 | 13.5 | 15 |
| Properties | Duration of Hydrothermal Synthesis at 200 °C—4 h | Curing Duration at 200 °C, h | |||
|---|---|---|---|---|---|
| 4 | 6 | 8 | 12 | ||
| Size of crystallites, nm | 35.0 | 38.1 | 39.9 | 43.1 | 50.3 |
| Average density, kg/m3 | – | 193.3 | 188.5 | 191.8 | 190.1 |
| Compressive strength, MPa | – | 0.77 | 0.87 | 0.92 | 0.93 |
| Bending strength, MPa | – | 0.46 | 0.56 | 0.53 | 0.59 |
| Properties | Duration of Synthesis at 220 °C, When Curing Duration at 220 °C—12 h | Curing Duration at 220 °C, When Duration of Synthesis at 220 °C—8 h | ||||
|---|---|---|---|---|---|---|
| Columns number | 1 | 2 | 3 | 4 | 5 | 6 |
| Hydrothermal treatment, h | 4 | 8 | 12 | 4 | 6 | 8 |
| Size of crystallites, nm | 30.4 | 31.7 | 34.0 | 27.3 | 27.9 | 30.7 |
| Average density, kg/m3 | 214.2 | 184.0 | 178,1 | 178.6 | 181.3 | 183.1 |
| Compressive strength, MPa | 1.40 | 1.54 | 1.84 | 1.40 | 1.34 | 1.44 |
| Bending strength, MPa | 0.78 | 0.83 | 0.86 | 0.61 | 0.68 | 0.85 |
| Properties | Burning Temperature, °C, With Duration of 2 h | ||
|---|---|---|---|
| 650 | 800 | 1000 | |
| Compressive strength, MPa | 1.39 | 1.22 | 0.97 |
| Variation, % of initial value | 96.5 | 84.7 | 67.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siauciunas, R.; Smalakys, G.; Eisinas, A.; Prichockiene, E. Synthesis of High Crystallinity 1.13 nm Tobermorite and Xonotlite from Natural Rocks, Their Properties and Application for Heat-Resistant Products. Materials 2022, 15, 3474. https://doi.org/10.3390/ma15103474
Siauciunas R, Smalakys G, Eisinas A, Prichockiene E. Synthesis of High Crystallinity 1.13 nm Tobermorite and Xonotlite from Natural Rocks, Their Properties and Application for Heat-Resistant Products. Materials. 2022; 15(10):3474. https://doi.org/10.3390/ma15103474
Chicago/Turabian StyleSiauciunas, R., G. Smalakys, A. Eisinas, and E. Prichockiene. 2022. "Synthesis of High Crystallinity 1.13 nm Tobermorite and Xonotlite from Natural Rocks, Their Properties and Application for Heat-Resistant Products" Materials 15, no. 10: 3474. https://doi.org/10.3390/ma15103474
APA StyleSiauciunas, R., Smalakys, G., Eisinas, A., & Prichockiene, E. (2022). Synthesis of High Crystallinity 1.13 nm Tobermorite and Xonotlite from Natural Rocks, Their Properties and Application for Heat-Resistant Products. Materials, 15(10), 3474. https://doi.org/10.3390/ma15103474