


A Novel Interstitial Site in Binary Rock-Salt Compounds



Abstract
:1. Introduction
2. Methods
3. Results
3.1. Energetic Stability of Neutral Defects
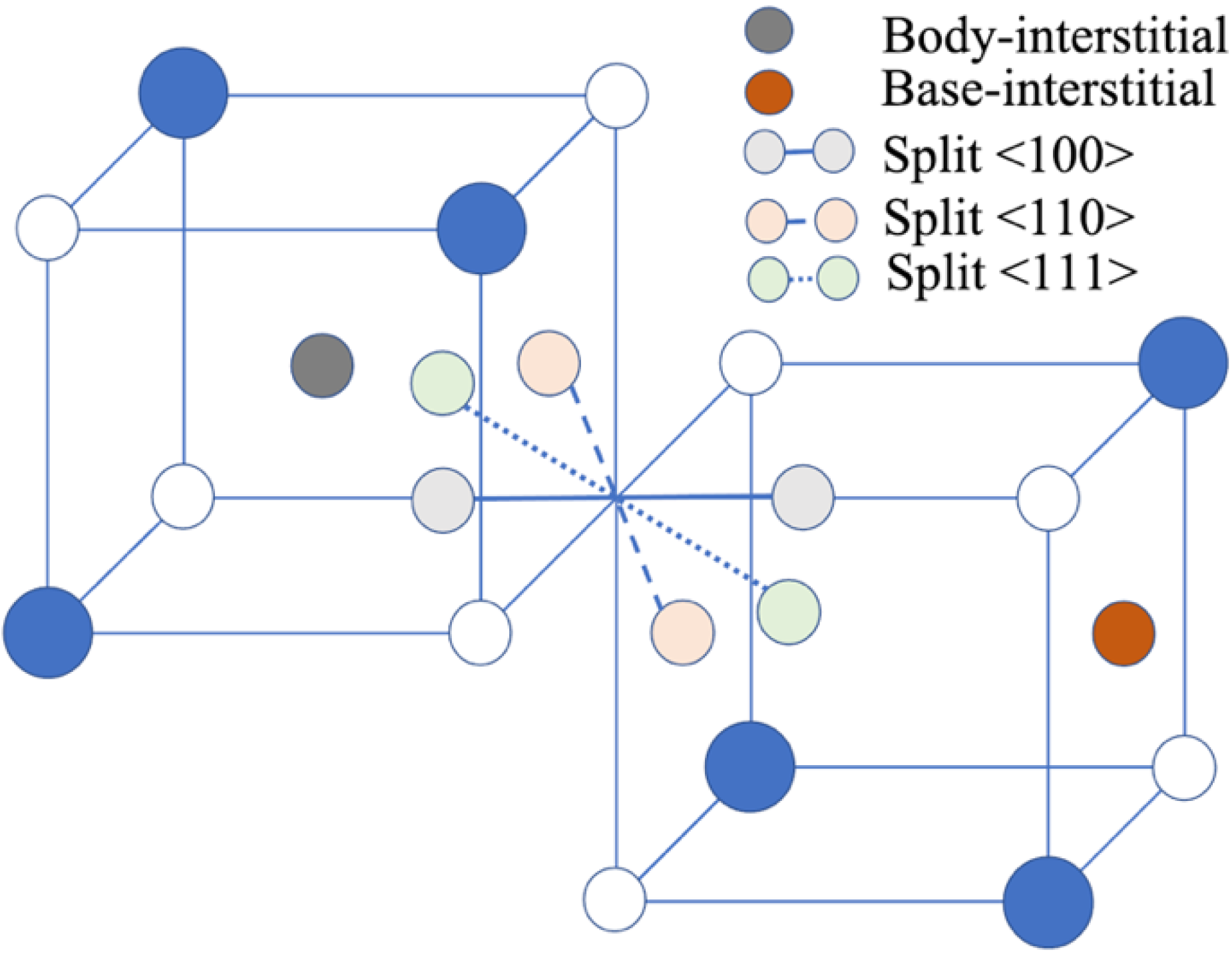
3.1.1. Neutral Anion Interstitial Point Defects
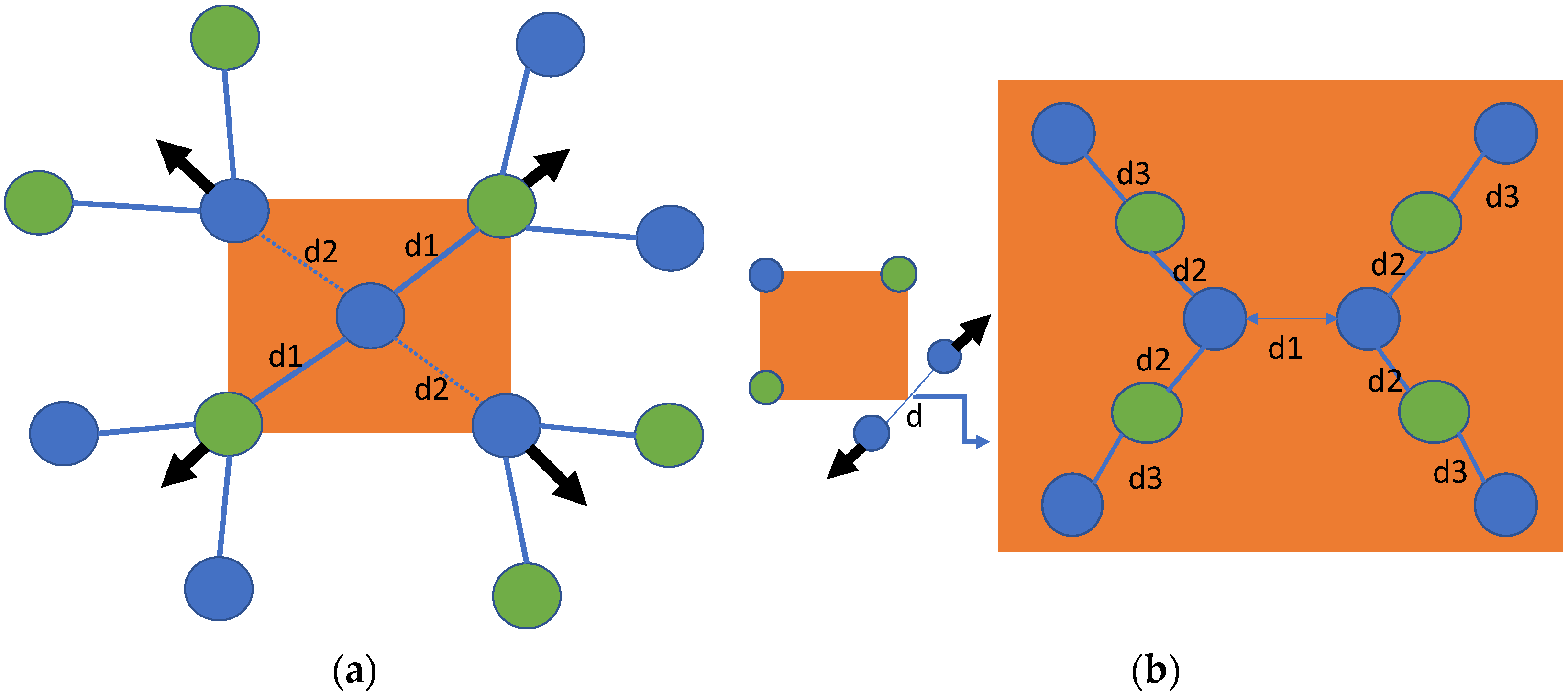
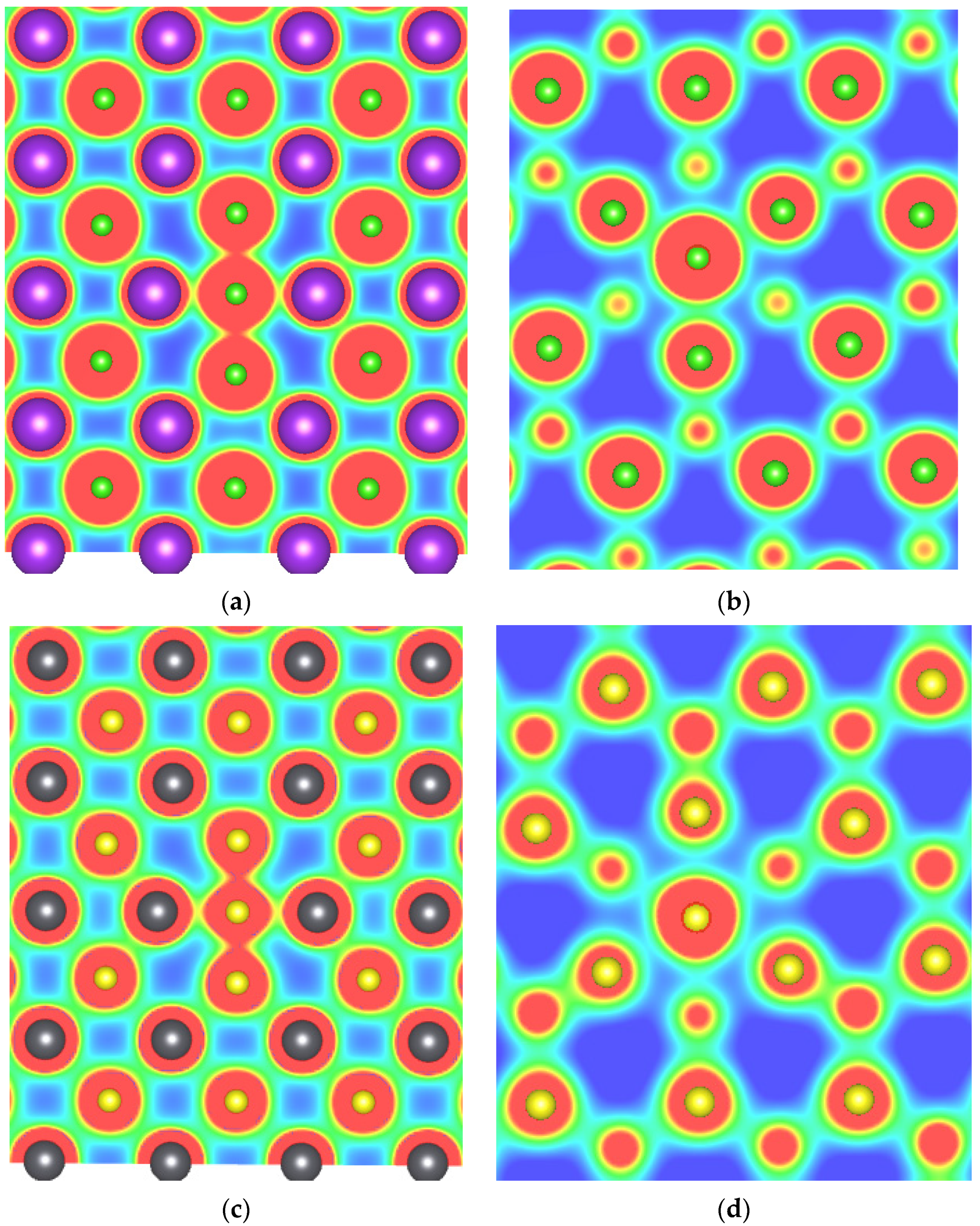
3.1.2. Crystal Structure Relaxation
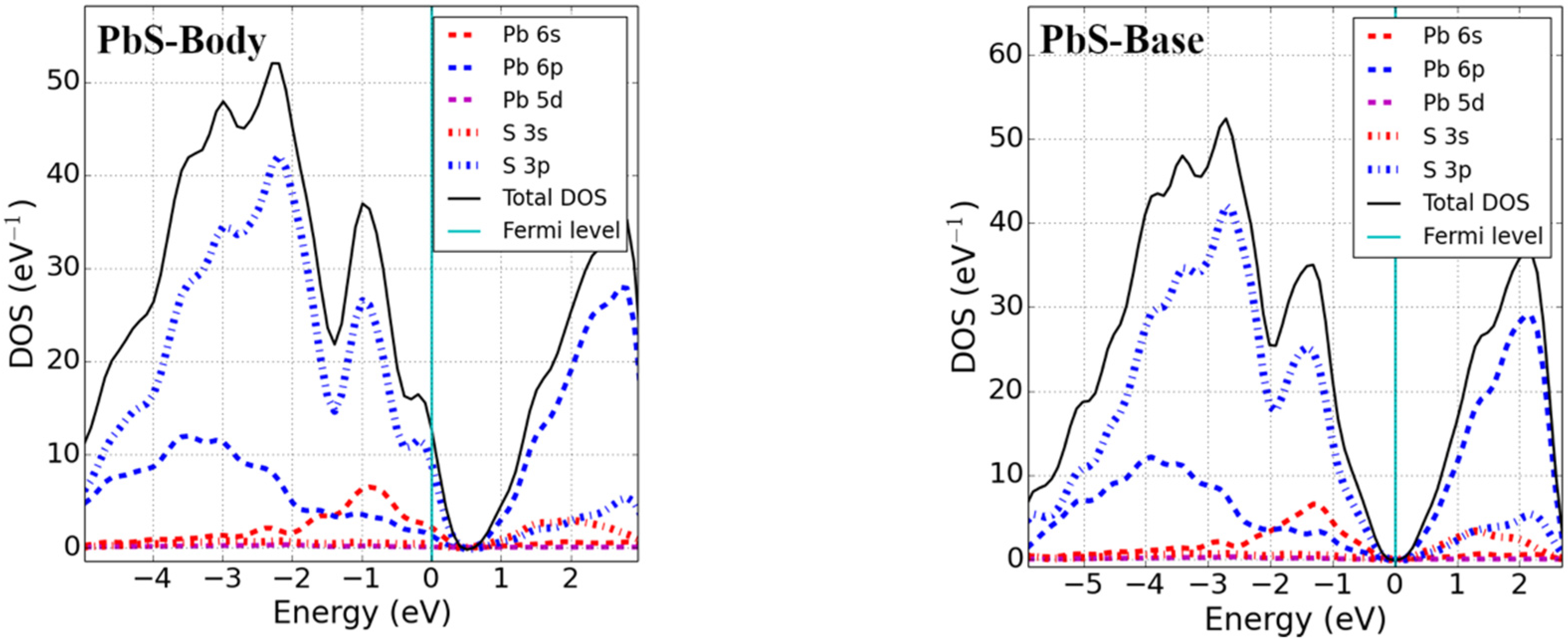
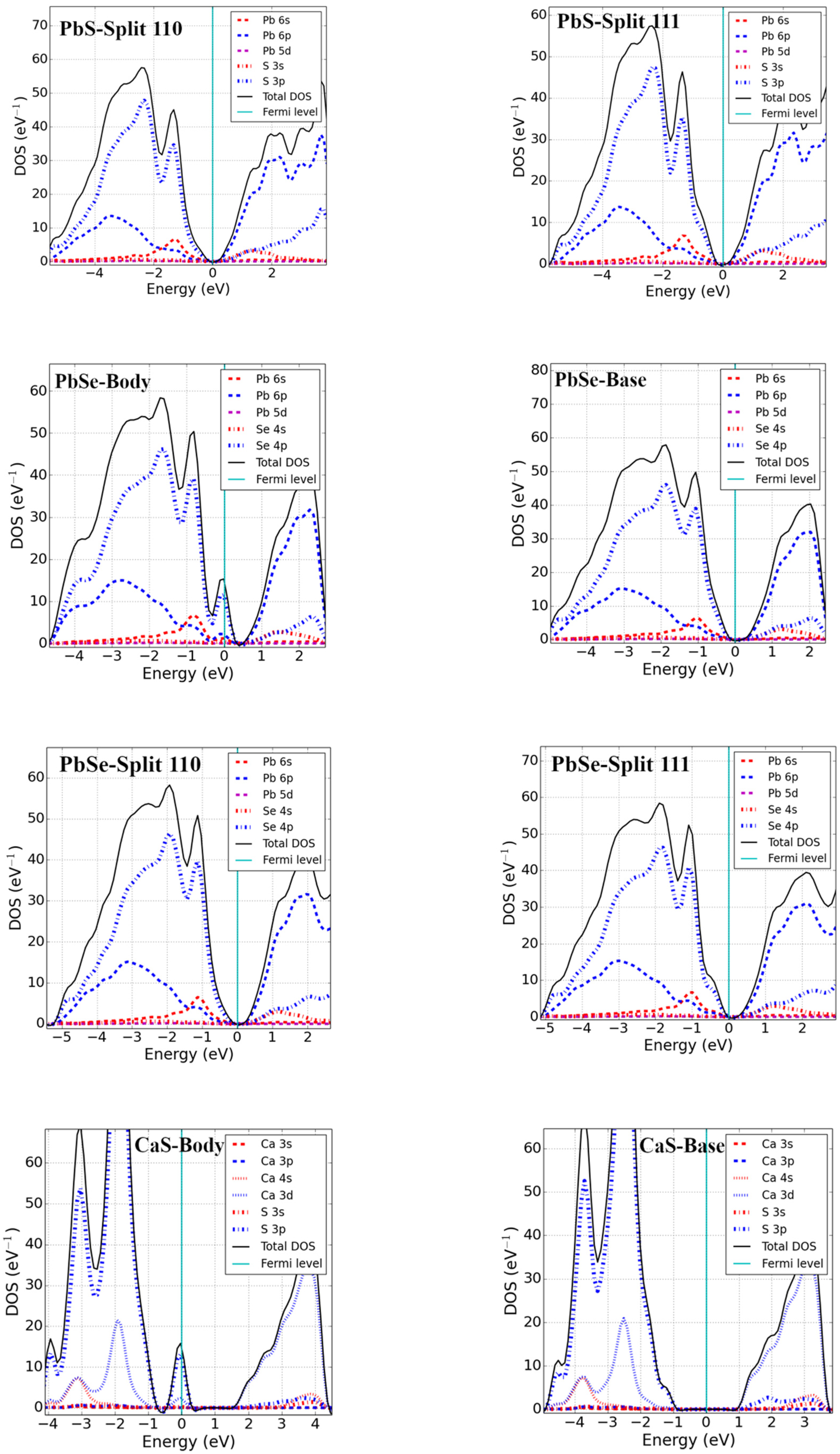
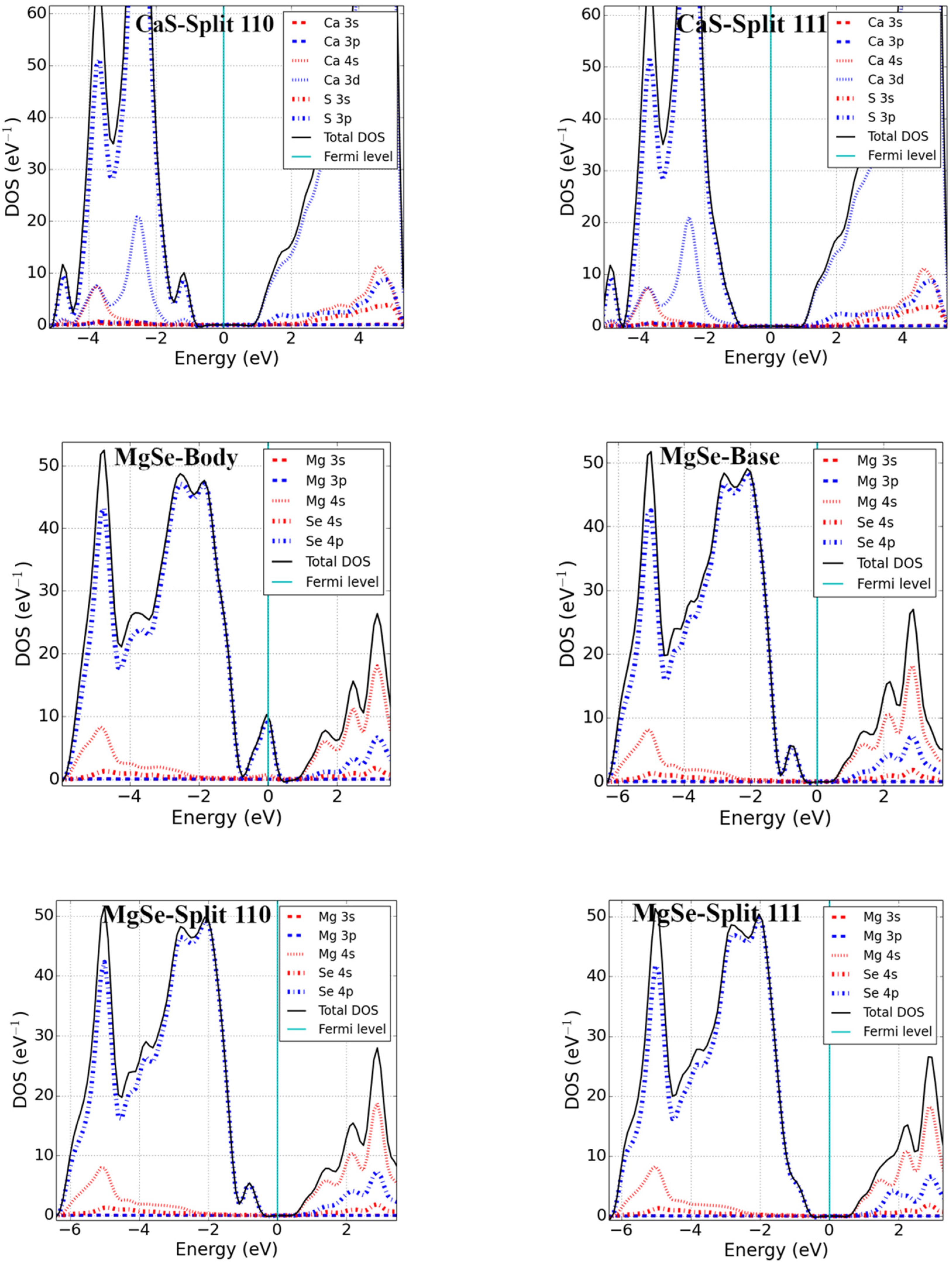
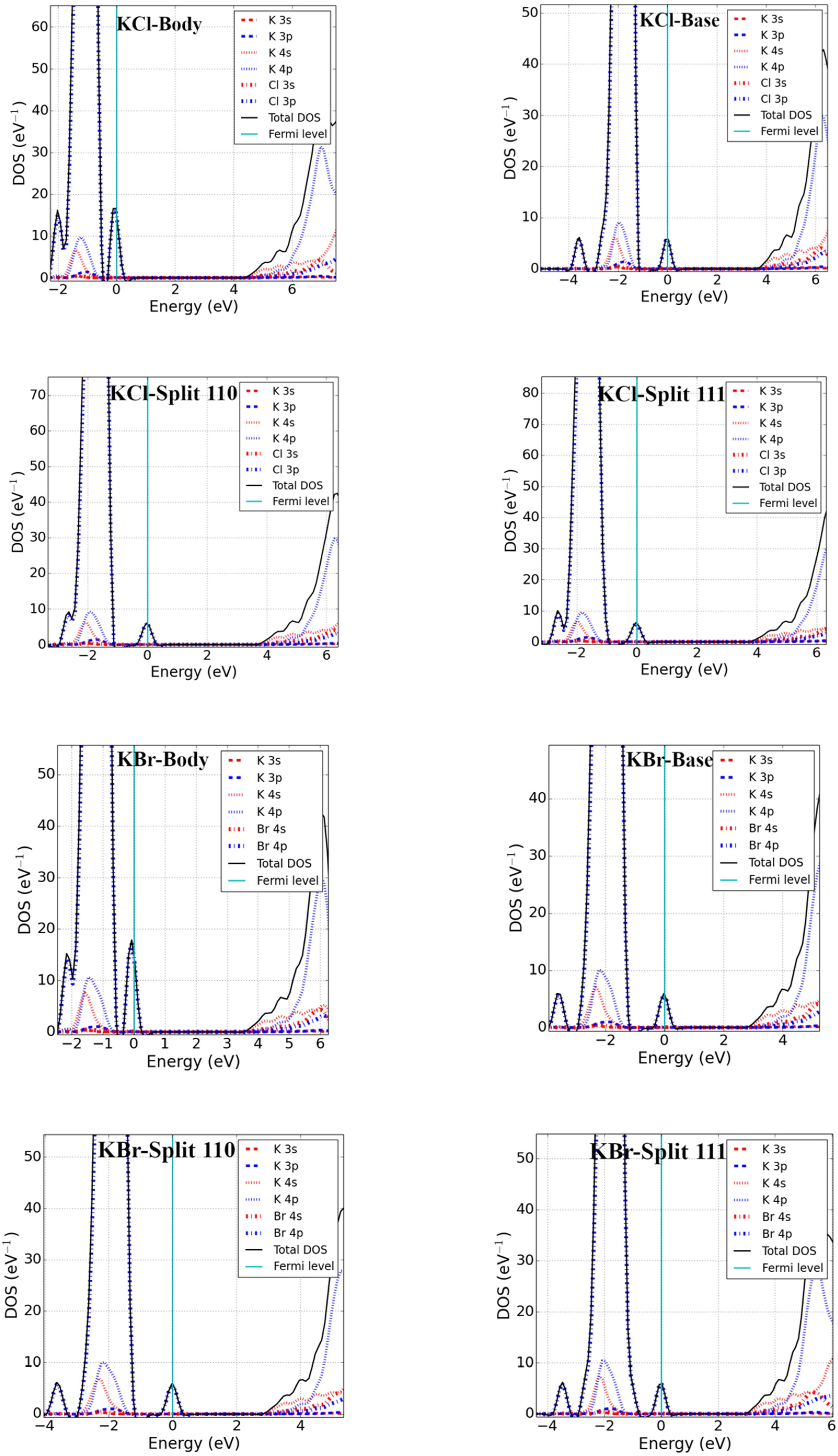
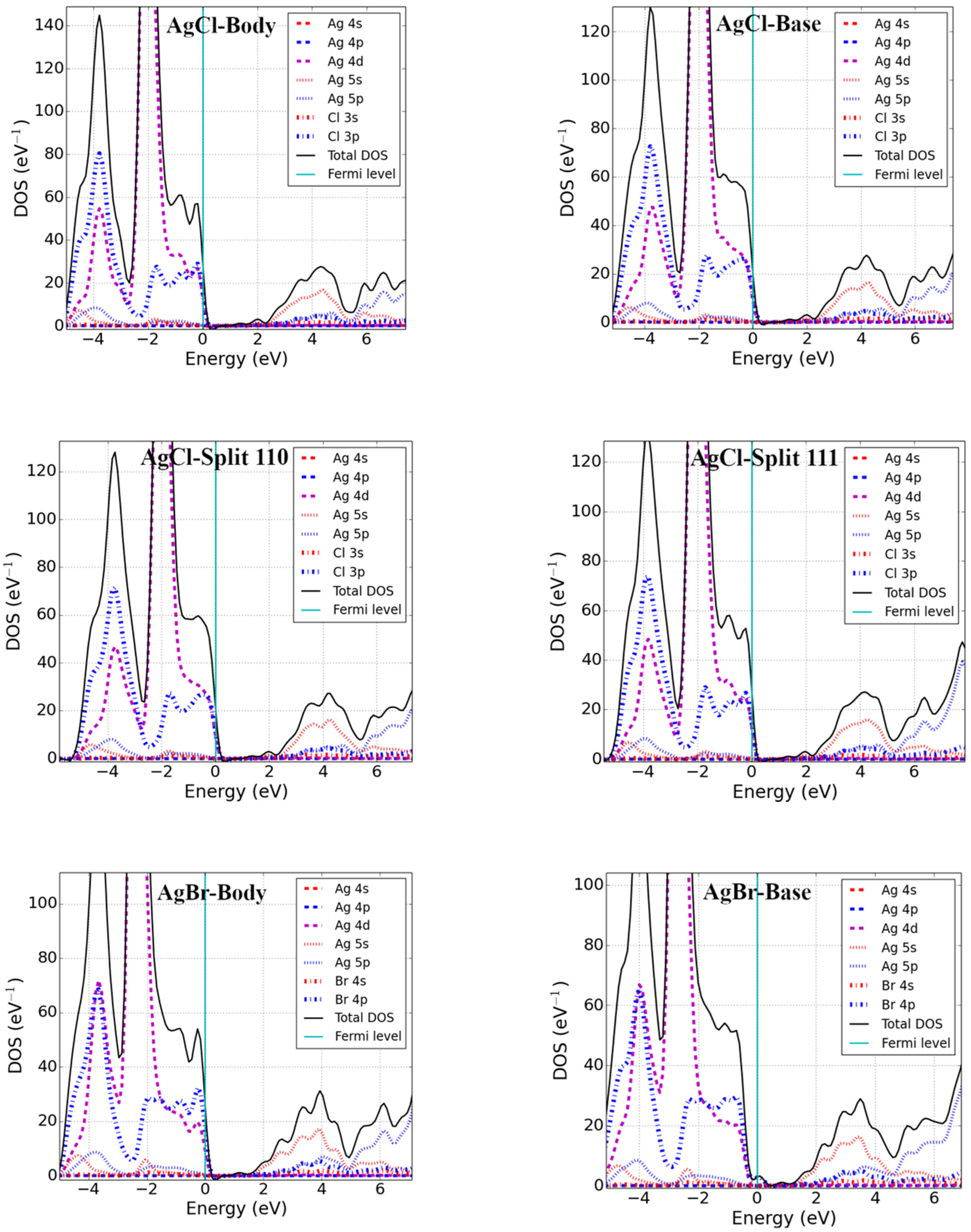
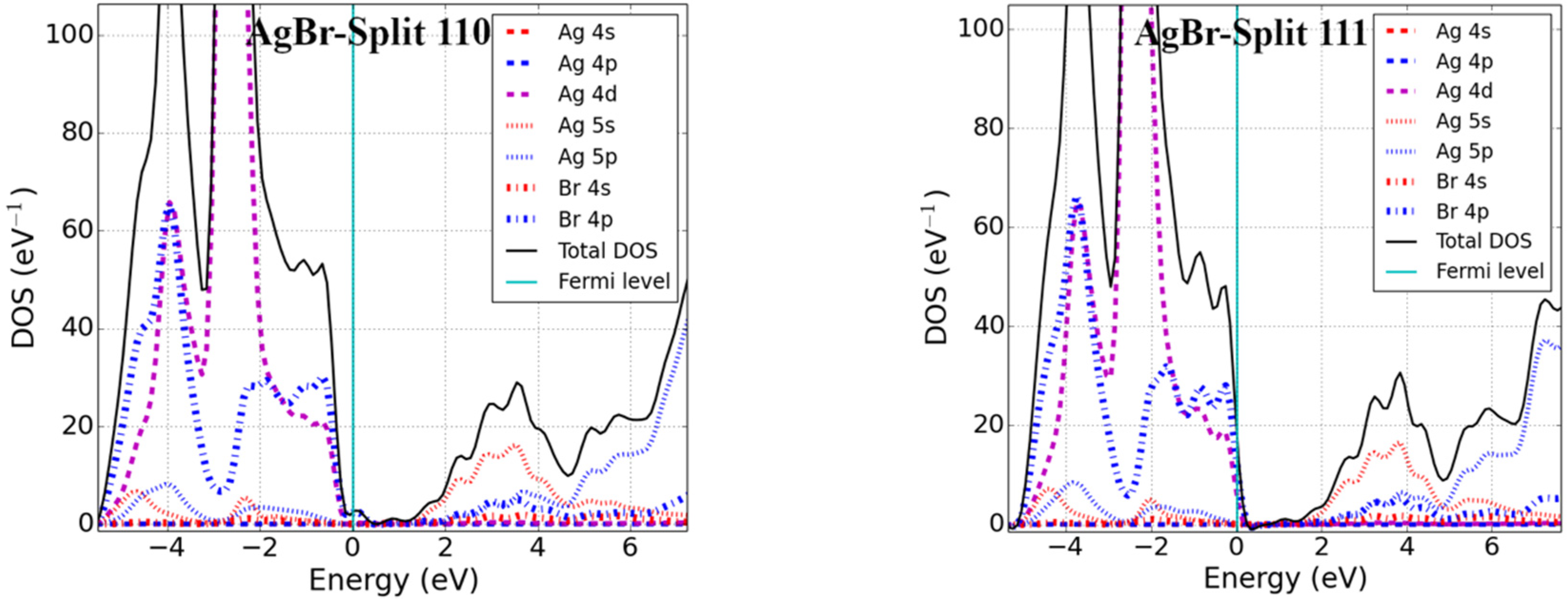
3.2. Bandgap and Electronic Properties of Neutral Defects
- (i)
- Pb monochalcogenides (PbX; X = S, Se)
- (ii)
- Alkaline-earth metal monochalcogenides (MX; M = Ca, Mg; X = S, Se)
- (iii)
- Potassium halides (KX′; X′ = Cl, Br)
- (iv)
- Silver halides (AgX′; X′ = Cl, Br)
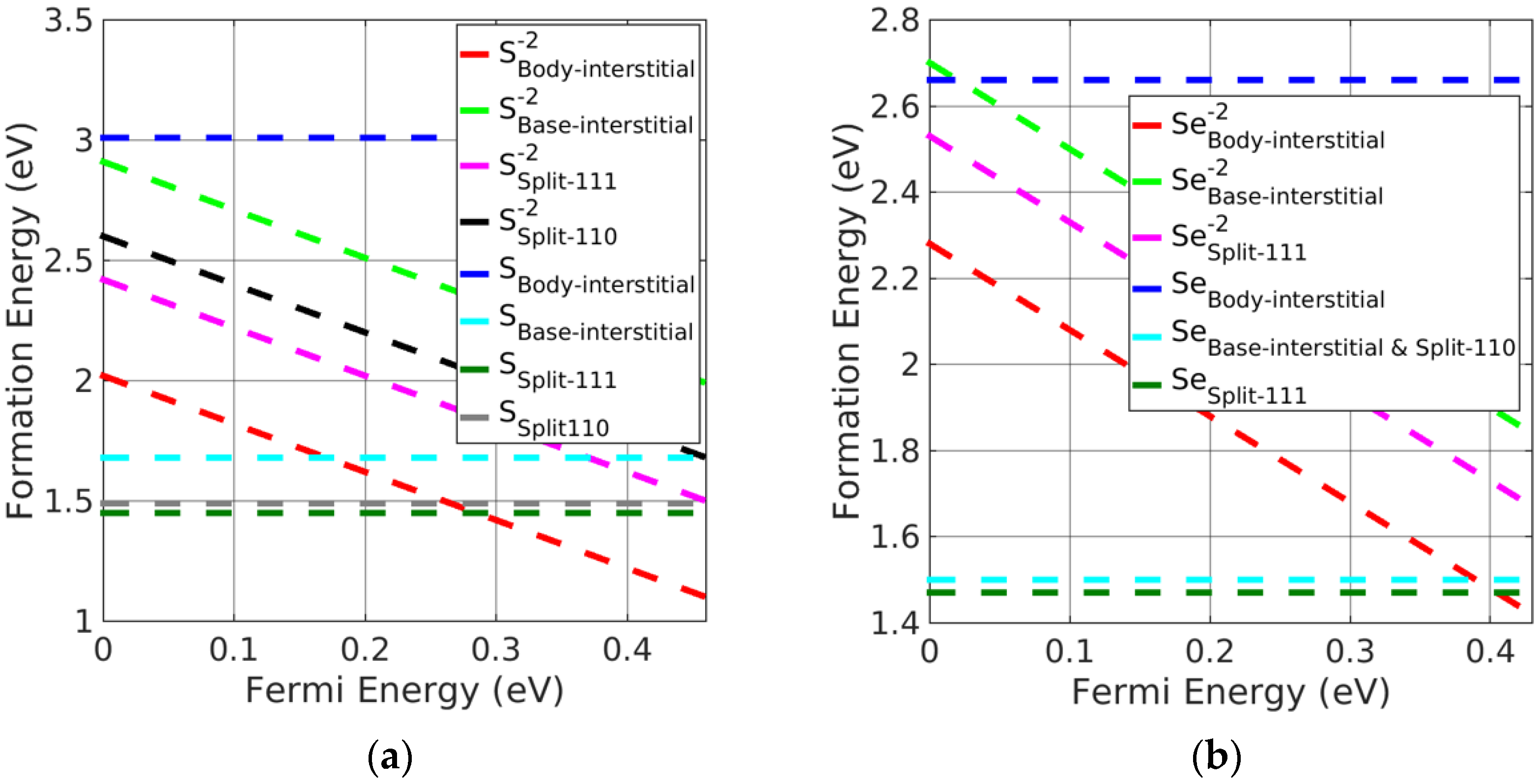
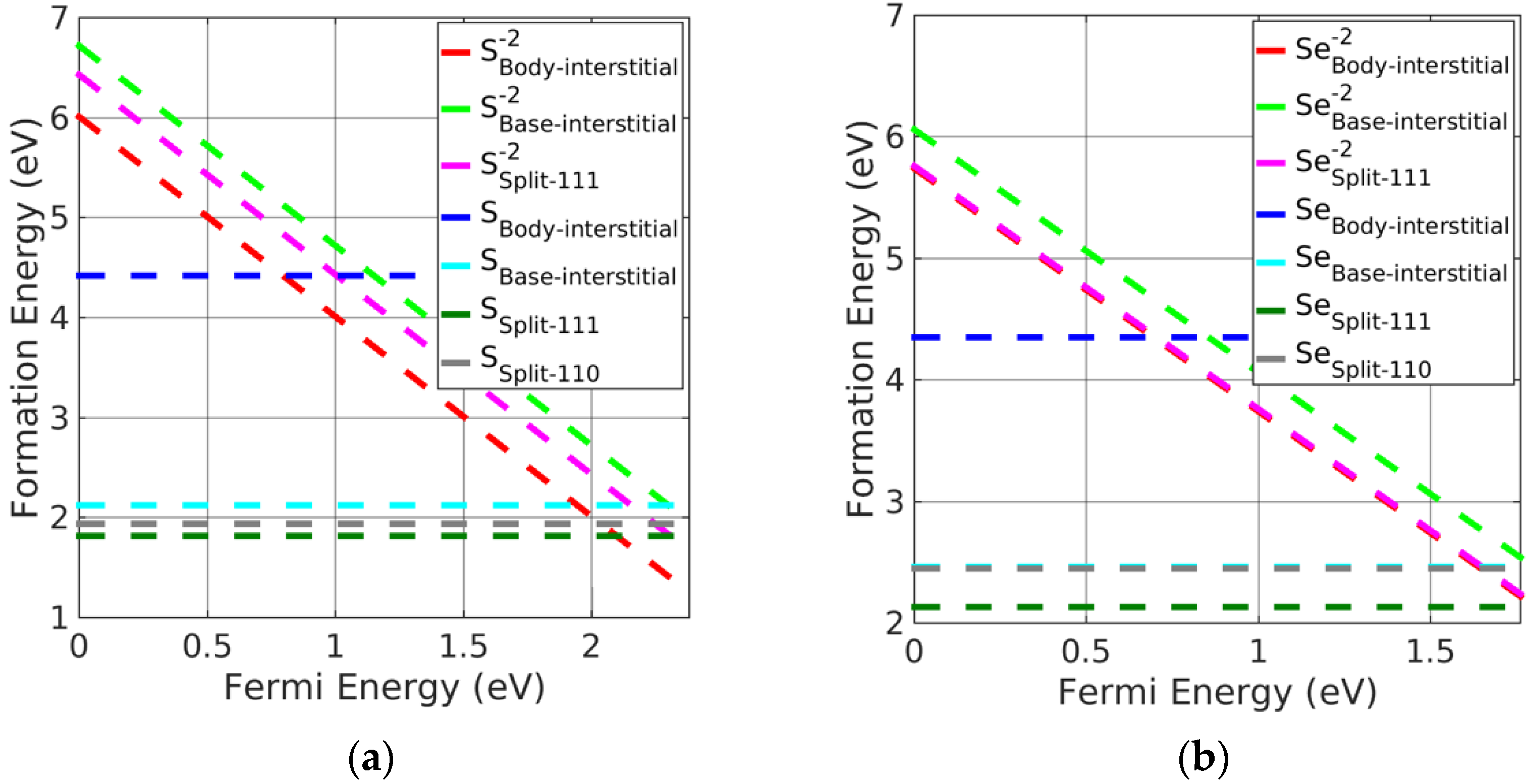
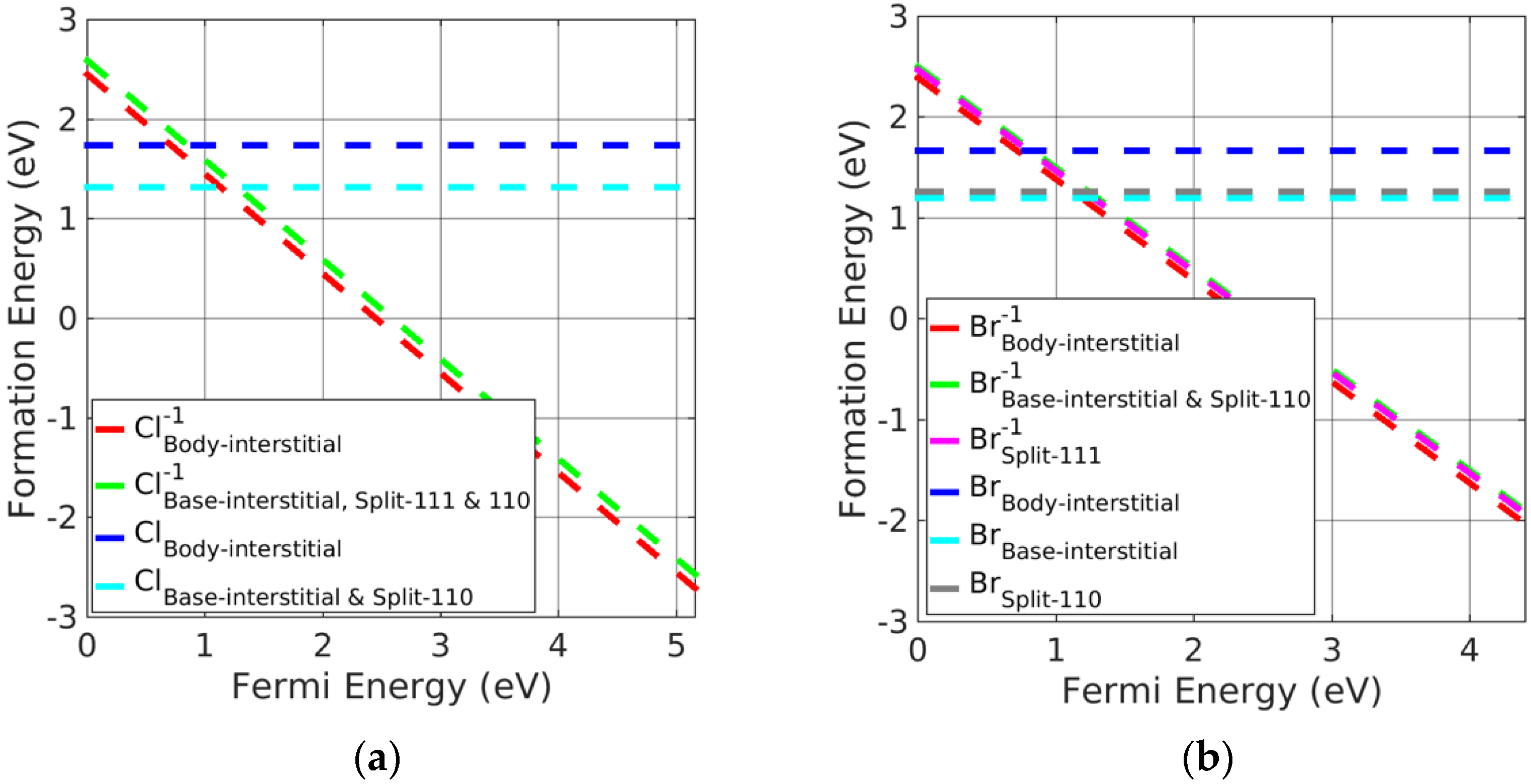
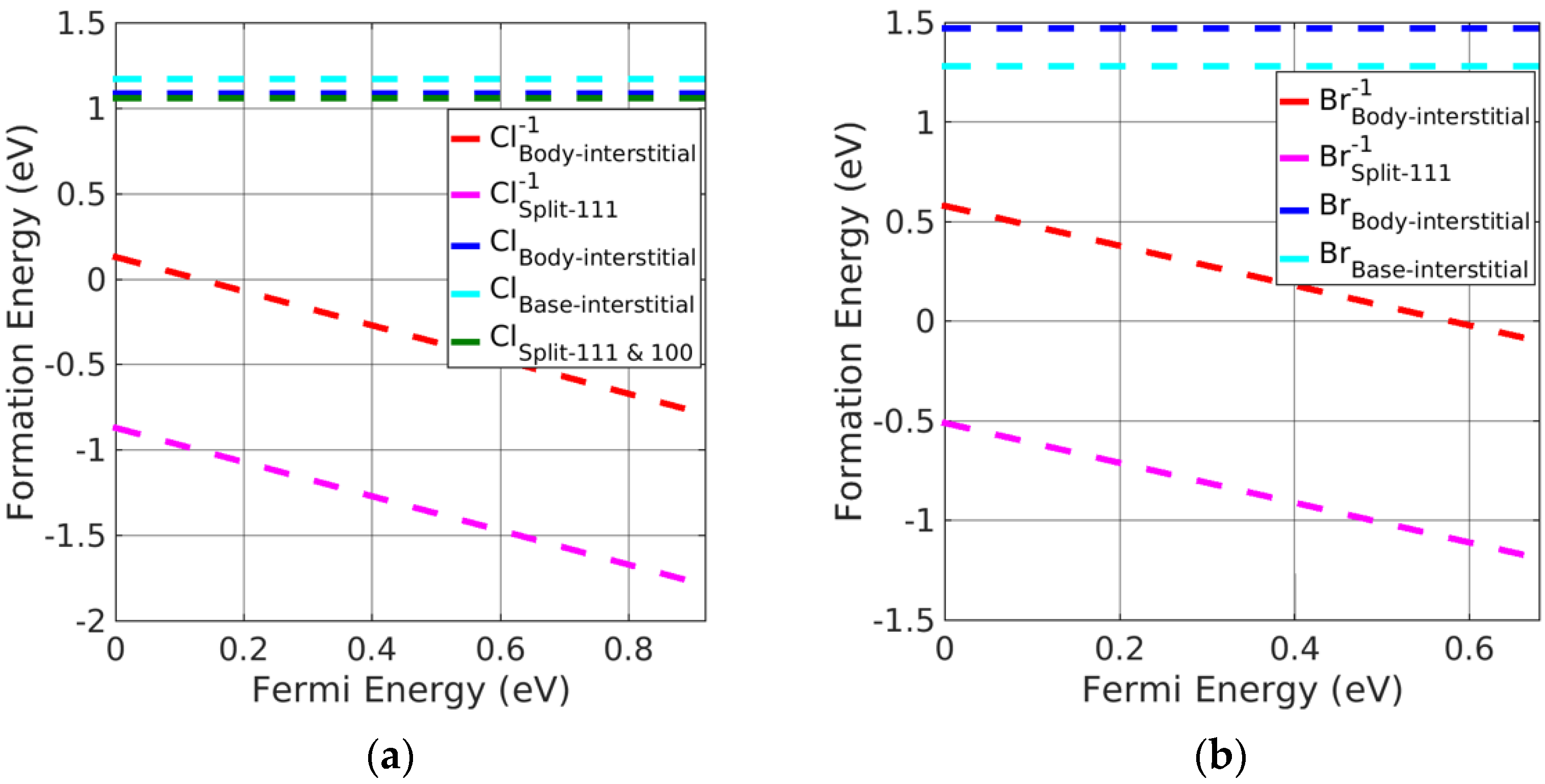
3.3. Investigation of Charged Interstitial Defects
- (i)
- Pb monochalcogenides
- (ii)
- Alkaline-earth metal monochalcogenides
- (iii)
- Potassium halides
- (iv)
- Silver halides
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; van de Walle, C.G. First-principles Calculations for Point Defects in Solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Lee, J.H.; Black, R.; Popov, G.; Pomerantseva, E.; Nan, F.; Botton, G.A.; Nazar, L.F. The Role of Vacancies and Defects in Na0.44MnO2 Nanowire Catalysts for Lithium–oxygen Batteries. Energy Environ. Sci. 2012, 5, 9558–9565. [Google Scholar] [CrossRef]
- Lu, I.T.; Bernardi, M. Using Defects to Store Energy in Materials – A Computational Study. Sci. Rep. 2017, 7, 3403. [Google Scholar] [CrossRef] [PubMed]
- Seitz, F. Color Centers in Alkali Halide Crystals. Rev. Mod. Phys. 1946, 18, 384–408. [Google Scholar] [CrossRef]
- Nan, H.; Wang, Z.; Wang, W.; Liang, Z.; Lu, Y.; Chen, Q.; He, D.; Tan, P.; Miao, F.; Wang, X.; et al. Strong Photoluminescence Enhancement of MoS2 through Defect Engineering and Oxygen Bonding. ACS Nano 2014, 8, 5738–5745. [Google Scholar] [CrossRef]
- Jiang, G.; He, J.; Zhu, T.; Fu, C.; Liu, X.; Hu, L.; Zhao, X. High Performance Mg2(Si,Sn) Solid Solutions: A Point Defect Chemistry Approach to Enhancing Thermoelectric Properties. Adv. Funct. Mater. 2014, 24, 3776–3781. [Google Scholar] [CrossRef]
- Yaremchenko, A.A.; Populoh, S.; Patricio, S.G.; Macias, J.; Thiel, P.; Fagg, D.P.; Weidenkaff, A.; Frade, J.R.; Kovalevsky, A.V. Boosting Thermoelectric Performance by Controlled Defect Chemistry Engineering in Ta-Substituted Strontium Titanate. Chem. Mater. 2015, 27, 4995–5006. [Google Scholar] [CrossRef]
- Suh, J.; Yu, K.M.; Fu, D.; Liu, X.; Yang, F.; Fan, J.; Smith, D.J.; Zhang, Y.H.; Furdyna, J.K.; Dames, C.; et al. Simultaneous Enhancement of Electrical Conductivity and Thermopower of Bi2Te3 by Multifunctionality of Native Defects. Adv. Mater. 2015, 27, 3681–3686. [Google Scholar] [CrossRef]
- Kobayashi, S.; Mizumukai, Y.; Ohnishi, T.; Shibata, N.; Ikuhara, Y.; Yamamoto, T. High Electron Mobility of Nb-Doped SrTiO3 Films Stemming from Rod-Type Sr Vacancy Clusters. ACS Nano 2015, 9, 10769–10777. [Google Scholar] [CrossRef]
- Kim, J.Y.; Oh, M.W.; Lee, S.; Cho, Y.C.; Yoon, J.H.; Lee, G.W.; Cho, C.R.; Park, C.H.; Jeong, S.Y. Abnormal Drop in Electrical Resistivity with Impurity Doping of Single-crystal Ag. Sci. Rep. 2014, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Claeys, C.; Simoen, E. Radiation Effects in Advanced Semiconductor Materials and Devices; Springer: Leuven, Belgium, 2002. [Google Scholar]
- West, A.R. Basic Solid State Chemistry and Its Applications, 2nd ed.; Wiley: Sheffield, UK, 2013. [Google Scholar]
- Li, H.; Huang, M.; Chen, S. First-principles Exploration of Defect-pairs in GaN. J. Semicond 2020, 41, 032104. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Khare, S.V.; Gall, D. Energetics of Point Defects in Rocksalt Structure Transition Metal Nitrides: Thermodynamic Reasons for Deviations from Stoichiometry. Acta Mater. 2018, 159, 77–88. [Google Scholar] [CrossRef]
- Tsetseris, L.; Kalfagiannis, N.; Logothetidis, S.; Pantelides, S.T. Structure and Interaction of Point Defects in Transition-metal Nitrides. Phys. Rev. B 2007, 76, 224107. [Google Scholar] [CrossRef]
- Gu, Z.; Hu, C.; Fan, X.; Xu, L.; Wen, M.; Meng, Q.; Zhao, L.; Zheng, X.; Zheng, W. On the Nature of Point Defect and its Effect on Electronic Structure of Rocksalt Hafnium Nitride Films. Acta Mater. 2014, 81, 315–325. [Google Scholar] [CrossRef]
- Moreno-Armenta, M.G.; Soto, G. Electronic Structure of Scandium Nitride with Nitrogen and Scandium Deficits. Comput. Mater. Sci. 2007, 40, 275–281. [Google Scholar] [CrossRef]
- Sun, W.; Ehteshami, H.; Korzhavyi, P.A. Structure and Energy of Point Defects in TiC: An ab initio Study. Phys. Rev. B 2015, 91, 134111. [Google Scholar] [CrossRef]
- Yang, G.; Xiong, M.; Zhou, Y.; Tao, X.; Peng, Q.; Ouyang, Y. The Effects of Temperature and Pressure on the Physical Properties and Stabilities of Point Defects and Defect Complexes in B1-ZrC. Comput. Mater. Sci. 2021, 198, 110694. [Google Scholar] [CrossRef]
- Tsetseris, L.; Pantelides, S.T. Vacancies, Interstitials and their Complexes in Titanium Carbide. Acta Mater. 2008, 56, 2864–2871. [Google Scholar] [CrossRef]
- Goyal, A.; Gorai, P.; Toberer, E.S.; Stevanović, V. First-principles Calculation of Intrinsic Defect Chemistry and Self-doping in PbTe. NPJ Comput. Mater. 2017, 3, 1–9. [Google Scholar] [CrossRef]
- Li, W.; Fang, C.; Dijkstra, M.; van Huis, M.A. The Role of Point Defects in PbS, PbSe and PbTe: A first principles Study. J. Phys. Condens. Matter. 2015, 27, 355801. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, Y.; Tsunoda, N.; Oba, F. Point Defects and p-Type Doping in ScN from First Principles. Phys. Rev. Appl. 2018, 9, 34019. [Google Scholar] [CrossRef]
- Rojas, T.; Ulloa, S.E. Energetics and Electronic Structure of Native Point Defects in Antiferromagnetic CrN. Phys. Rev. B 2018, 98, 214111. [Google Scholar] [CrossRef]
- Huang, B. Native Point Defects in CaS: Focus on Intrinsic Defects and Rare Earth Ion Dopant Levels for Up-converted Persistent Luminescence. Inorg. Chem. 2015, 54, 11423–11440. [Google Scholar] [CrossRef] [PubMed]
- Häfner, M.; Bredow, T. F and M Centers in Alkali Halides: A Theoretical Study Applying Self-Consistent Dielectric-Dependent Hybrid Density Functional Theory. Phys. Rev. B 2020, 102, 184108. [Google Scholar] [CrossRef]
- Kuzovkov, V.N.; Popov, A.I.; Kotomin, E.A.; Moskina, A.M.; Vasilchenko, E.; Lushchik, A. Theoretical Analysis of the Kinetics of Low-Temperature Defect Recombination in Alkali Halide Crystals. Low Temp. Phys. 2016, 42, 588–593. [Google Scholar] [CrossRef]
- Svane, A.; Kotomin, E.; Christensen, N.E. First-Principles Calculations of the Vibrational Properties of H Centers in KCl Crystals. Phys. Rev. B 1996, 53, 24–27. [Google Scholar] [CrossRef]
- Stevens, F.; Vrielinck, H.; van Speybroeck, V.; Pauwels, E.; Callens, F.; Waroquier, M. X- (X = O, S) Ions in Alkali Halide Lattices through Density Functional Calculations. 1. Substitutional Defect Models. J. Phys. Chem. B 2006, 110, 8204–8212. [Google Scholar] [CrossRef]
- di Valentin, C.; Pacchioni, G. g-tensor and Hyperfine Splitting of , , , Defect Centers in KCl from DFT Calculations. Model. Simul. Mater. Sci. Eng. 2009, 17, 084005. [Google Scholar] [CrossRef]
- Häfner, M.; Bredow, T. Mobility of F Centers in Alkali Halides. J. Phys. Chem. C 2021, 125, 9085–9095. [Google Scholar] [CrossRef]
- Lushchik, A.; Lushchik, C.; Vasil’Chenko, E.; Popov, A.I. Radiation Creation of Cation Defects in Alkali Halide Crystals: Review and Today’s Concept (Review Article). Low Temp. Phys. 2018, 44, 269–277. [Google Scholar] [CrossRef]
- Estreicher, S.K.; Backlund, D.; Gibbon, T.M.; Doçaj, A. Vibrational Properties of Impurities in Semiconductors. Model. Simul. Mater. Sci. Eng. 2009, 17, 084006. [Google Scholar] [CrossRef]
- Wilson, D.J.; Sokol, A.A.; French, S.A.; Catlow, C.R.A. Defect Structures in the Silver Halides. Phys. Rev. B 2008, 77, 064115. [Google Scholar] [CrossRef]
- Wilson, D.J.; Sokol, A.A.; French, S.A.; Catlow, C.R.A. Defect Structures in Silver Chlorides. J. Phys. Condens. Matter 2004, 16, S2827. [Google Scholar] [CrossRef]
- Mishra, N.; Makov, G. Point Defects in Lead Sulfide: A First-Principles Study. Comput. Mater. Sci. 2021, 190, 110285. [Google Scholar] [CrossRef]
- Bajaj, S.; Pomrehn, G.S.; Doak, J.W.; Gierlotka, W.; Wu, H.J.; Chen, S.W.; Wolverton, C.; Goddard, W.A., III; Snyder, G.J. Ab initio Study of Intrinsic Point Defects in PbTe: An Insight into Phase Stability. Acta Mater. 2015, 92, 72–80. [Google Scholar] [CrossRef]
- Gilbert, C.A.; Kenny, S.D.; Smith, R.; Sanville, E. Ab initio Study of Point Defects in Magnesium Oxide. Phys. Rev. B 2007, 76, 184103. [Google Scholar] [CrossRef]
- Mahmoud, S.; Carrez, P.; Reis, M.L.D.; Mousseau, N.; Cordier, P. Diffusion Mechanism of Bound Schottky Defect in Magnesium Oxide. Phys. Rev. Mater. 2021, 5, 33609. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for High-Throughput DFT Calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Erratum: “Hybrid Functionals Based on a Screened Coulomb Potential” [J. Chem. Phys. 118, 8207 (2003)]. J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Madelung, O. Semiconductors: Group IV Elements, IV–IV and III–IV Compounds (Landolt-Börnstein, New Series, Group III); Springer: Berlin, Germany, 2005; Volume 41. [Google Scholar]
- Strehlow, W.H.; Cook, E.L. Compilation of Energy Band Gaps in Elemental and Binary Compound Semiconductors and Insulators. J. Phys. Chem. Ref. Data 1973, 2, 163–200. [Google Scholar] [CrossRef]
- Glascock, H.H.; Hensley, E.B. Fundamental Optical Absorption, Electrical Conductivity, and Thermoelectric Power of Calcium Oxide. Phys. Rev. 1963, 131, 649–652. [Google Scholar] [CrossRef]
- Luo, H.; Greene, R.G.; Ghandehari, K.; Li, T.; Ruoff, A.L. Structural Phase Transformations and the Equations of State of Calcium Chalcogenides at High Pressure. Phys. Rev. B 1994, 50, 16232–16237. [Google Scholar] [CrossRef] [PubMed]
- Gopikrishnan, C.R.; Jose, D.; Dattaa, A. Electronic Structure, Lattice Energies and Born Exponents for Alkali Halides from First Principles. AIP Adv. 2012, 2, 012131. [Google Scholar] [CrossRef]
- Brown, F.C.; Gahwiller, C.; Fujita, H.; Kunz, A.B.; Scheifley, W.; Carrerat, N. Extreme-Ultraviolet Spectra of Ionic Crystals. Phys. Rev. B 1970, 2, 2126. [Google Scholar] [CrossRef]
- Okoye, C.M.I. Full-Potential Study of the Electronic Structure of Silver Halides. Phys. Status Solidi B 2002, 234, 580–589. [Google Scholar] [CrossRef]
- Smith, P.V. A Tight-Binding Approach to the Electronic Structure of the Silver Halides—II: The Silver Iodide Polymorphs. J. Phys. Chem. Solids 1976, 37, 589–597. [Google Scholar] [CrossRef]
- Biunno, N.; Narayan, J.; Hofmeister, S.K.; Srivatsa, A.R.; Singh, R.K. Low-Temperature Processing of Titanium Nitride Films by Laser Physical Vapor Deposition. Appl. Phys. Lett. 1989, 54, 1519–1521. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Lattice Constant (Å) | Experimental Lattice Constant (Å) | Bandgap (eV) | Experimental Bandgap at 300 K (eV) |
|---|---|---|---|---|
| PbS | 5.981 | 5.936 [45] | 0.46 | 0.41 [46] |
| PbSe | 6.215 | 6.124 [45] | 0.43 | 0.29 [46] |
| PbTe | 6.562 | 6.462 [45] | 0.80 | 0.32 [46] |
| CaO | 4.831 | 4.811 [45] | 3.67 | 7.70 [47] |
| CaS | 5.71 | 5.69 [48] | 2.38 | 5.80 [46] |
| MgSe | 5.508 | 5.46 [45] | 1.76 | 5.6 [46] |
| NaCl | 5.705 | 5.64 [49] | 4.98 | 8.75 [49] |
| NaBr | 6.039 | 5.977 [49] | 4.06 | 7.10 [49] |
| NaI | 6.533 | 6.473 [49] | 3.57 | 5.90 [50] |
| KCl | 6.308 | 6.293 [49] | 5.16 | 8.4 [49] |
| KBr | 6.626 | 6.597 [49] | 4.40 | 7.4 [49] |
| KI | 7.084 | 7.066 [49] | 3.95 | 6.0 [49] |
| RbBr | 7.012 | 6.889 [49] | 4.20 | 7.5 [49] |
| RbI | 7.487 | 7.342 [49] | 3.77 | 6.2 [49] |
| AgCl | 5.581 | 5.55 [51] | 0.92 | 3.25 [52] |
| AgBr | 5.812 | 5.77 [34] | 0.68 | 2.68 [52] |
| AgI | 6.128 | 6.07 [51] | 0.68 | 2.33 [52] |
| TiN | 4.247 | 4.25 [53] |
| Lead Monochalcogenides | |||
|---|---|---|---|
| Defect | PbS | PbSe | PbTe |
| Formation energy | Formation energy | Formation energy | |
| Base-interstitial | 1.68 | 1.5 | 1.82 |
| Body-interstitial | 3.01 | 2.66 | 2.69 |
| Split <111> | 1.45 | 1.47 | 1.76 |
| Split <110> | 1.49 | 1.50 | 1.82 |
| Split <100> | 2.13 | 2.39 | 3.14 |
| Alkaline-Earth Metal Monochalcogenides | |||
| MgSe | CaO | CaS | |
| Formation energy | Formation energy | Formation energy | |
| Base-interstitial | 2.46 | 2.04 | 2.12 |
| Body-interstitial | 4.35 | 3.56 | 4.42 |
| Split <111> | 2.13 | 1.31 | 1.82 |
| Split <110> | 2.45 | 1.18 | 1.94 |
| Split <100> | 4.82 | 2.93 | |
| Potassium Halides | |||
| KCl | KBr | KI | |
| Formation energy | Formation energy | Formation energy | |
| Base-interstitial | 1.32 | 1.2 | 1.04 |
| Body-interstitial | 1.74 | 1.67 | 1.55 |
| Split <111> | 1.41 | 1.31 | 1.15 |
| Split <110> | 1.32 | 1.26 | 1.06 |
| Split <100> | 1.73 | ||
| Sodium Halides | |||
| NaCl | NaBr | NaI | |
| Formation energy | Formation energy | Formation energy | |
| base-interstitial | 1.71 | 1.55 | 1.41 |
| body-interstitial | 2.3 | 2.16 | 2.01 |
| split <111> | 1.82 | 1.69 | 1.55 |
| split <110> | 1.71 | 1.56 | 1.41 |
| split <100> | 2.38 | 2.28 | |
| Rubidium Halides | |||
| RbBr | RbI | ||
| Formation energy | Formation energy | ||
| Base-interstitial | 1.03 | 0.90 | |
| Body-interstitial | 1.43 | 1.34 | |
| Split <111> | 1.13 | 0.99 | |
| Split <110> | 1.05 | 0.91 | |
| Split <100> | 1.37 | ||
| Silver Halides | |||
| AgCl | AgBr | AgI | |
| Formation energy | Formation energy | Formation energy | |
| Base-interstitial | 1.15 | 1.28 | 1.33 |
| Body-interstitial | 1.09 | 1.47 | 1.75 |
| Split <111> | 1.06 | 1.31 | 1.11 |
| Split <110> | 1.21 | 1.34 | 1.34 |
| Split <100> | 1.06 | 1.49 | 1.79 |
| Transition-Metal Nitride | |||
| TiN | |||
| Formation energy | |||
| Base-interstitial | 4.08 | ||
| Body-interstitial | 4.83 | ||
| Split <111> | 4.27 | ||
| Split <110> | 4.08 | ||
| Split <100> | 4.98 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, N.; Makov, G. A Novel Interstitial Site in Binary Rock-Salt Compounds. Materials 2022, 15, 6015. https://doi.org/10.3390/ma15176015
Mishra N, Makov G. A Novel Interstitial Site in Binary Rock-Salt Compounds. Materials. 2022; 15(17):6015. https://doi.org/10.3390/ma15176015
Chicago/Turabian StyleMishra, Neeraj, and Guy Makov. 2022. "A Novel Interstitial Site in Binary Rock-Salt Compounds" Materials 15, no. 17: 6015. https://doi.org/10.3390/ma15176015
APA StyleMishra, N., & Makov, G. (2022). A Novel Interstitial Site in Binary Rock-Salt Compounds. Materials, 15(17), 6015. https://doi.org/10.3390/ma15176015