
Dicalcium Phosphate Dihydrate Mineral Loaded Freeze-Dried Scaffolds for Potential Synthetic Bone Applications

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
- (1)
- Dicalcium Phosphate Dihydrate Mineral (DCPD)
- (2)
- Chitosan (CS) Stock Solution
- (3)
- Unloaded and DCPD Mineral Loaded Chitosan Scaffolds
- (4)
- Alkaline Treatment
2.1. Characterisation Techniques
2.1.1. Fourier Transform Infrared Spectroscopy (FTIR)
2.1.2. X-ray Diffraction (XRD)
2.1.3. Scanning Electron Microscopy (SEM)
2.2. Testing Techniques
2.2.1. Simultaneous Thermal Analysis (STA)
2.2.2. Mechanical Testing
2.2.3. Scaffold Swelling
2.2.4. Degradation Stability Testing
2.2.5. Zeta Potential
2.3. In Vitro Testing
2.3.1. Contact Cytotoxicity Assay by Giemsa Staining
2.3.2. Fluorescence Actin and Nuclei Staining
2.3.3. Extract Cytotoxicity by XTT Assay
2.3.4. DNA Quantification by Picogreen Assay
2.4. Statistical Analysis
3. Results
3.1. Fourier Transform Infrared Spectroscopy
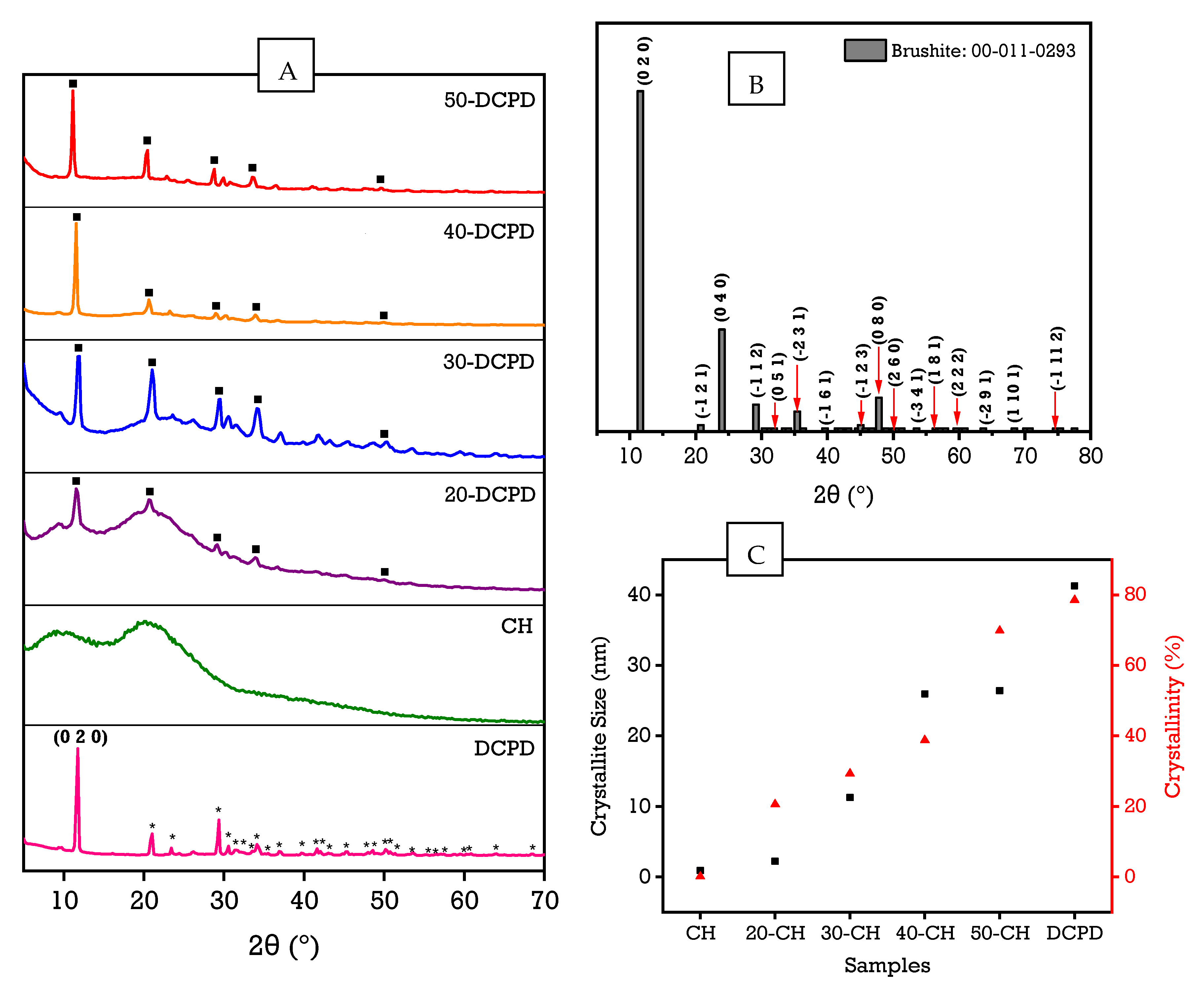
3.2. X-ray Diffraction
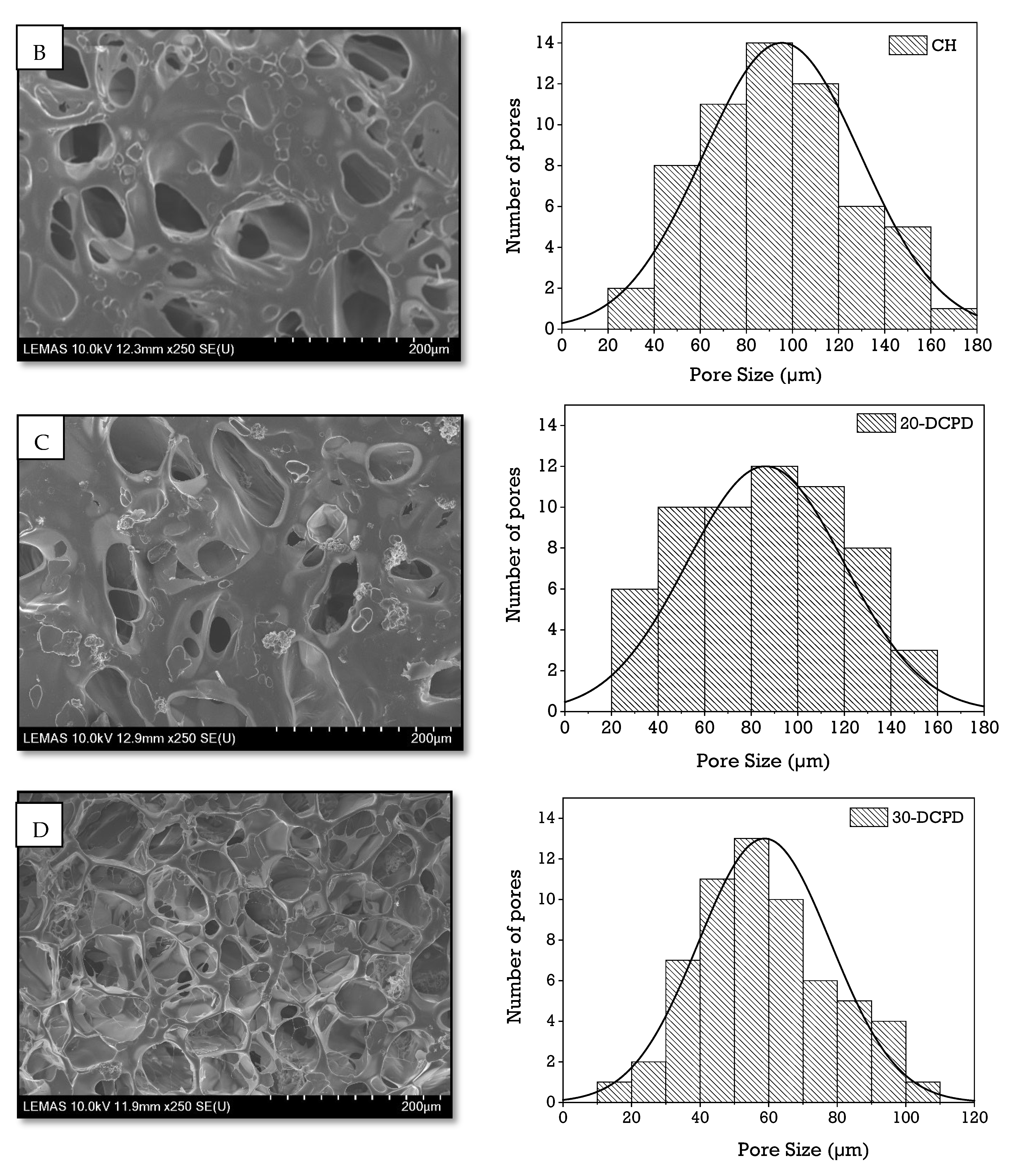
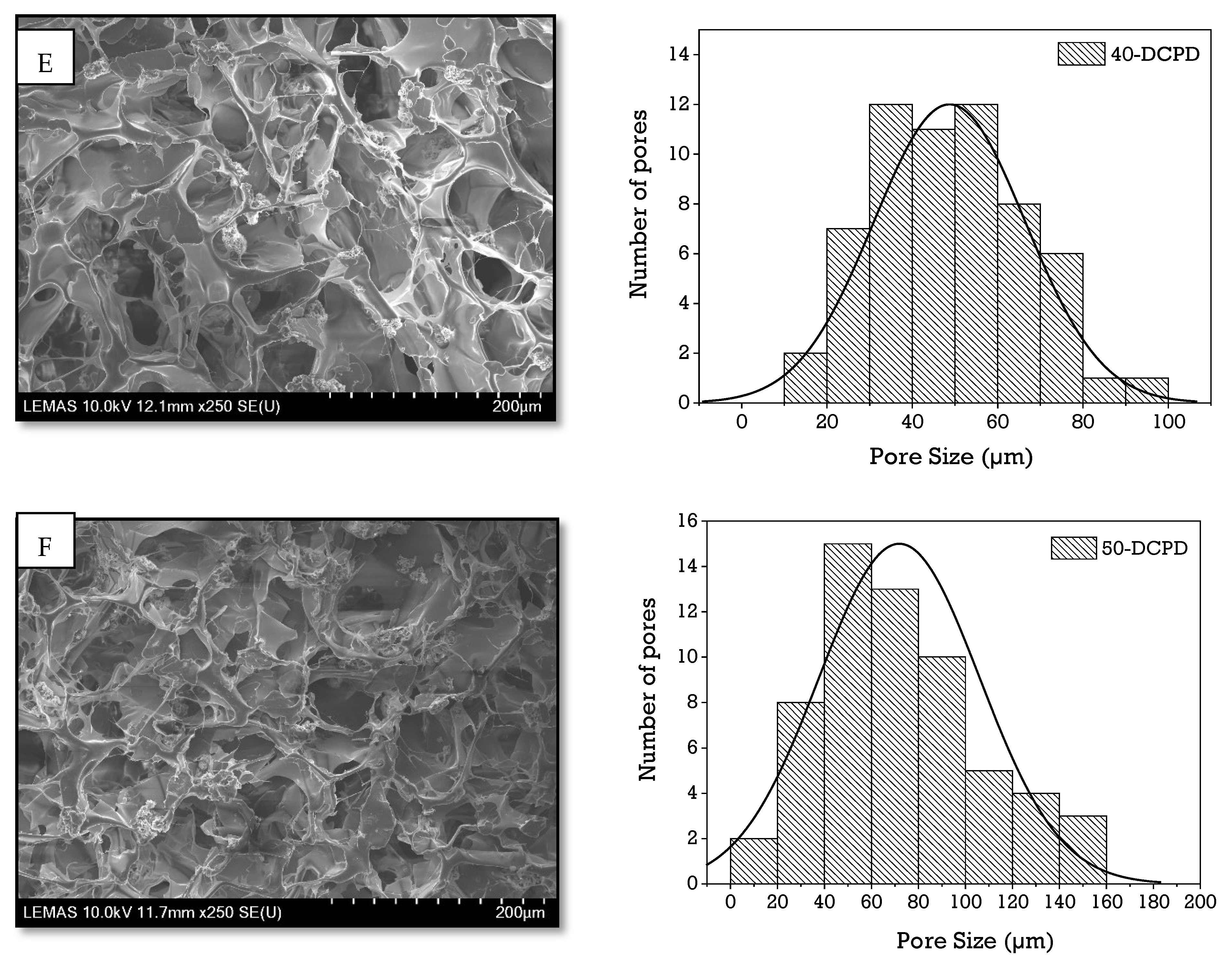
3.3. Scanning Electron Microscopy
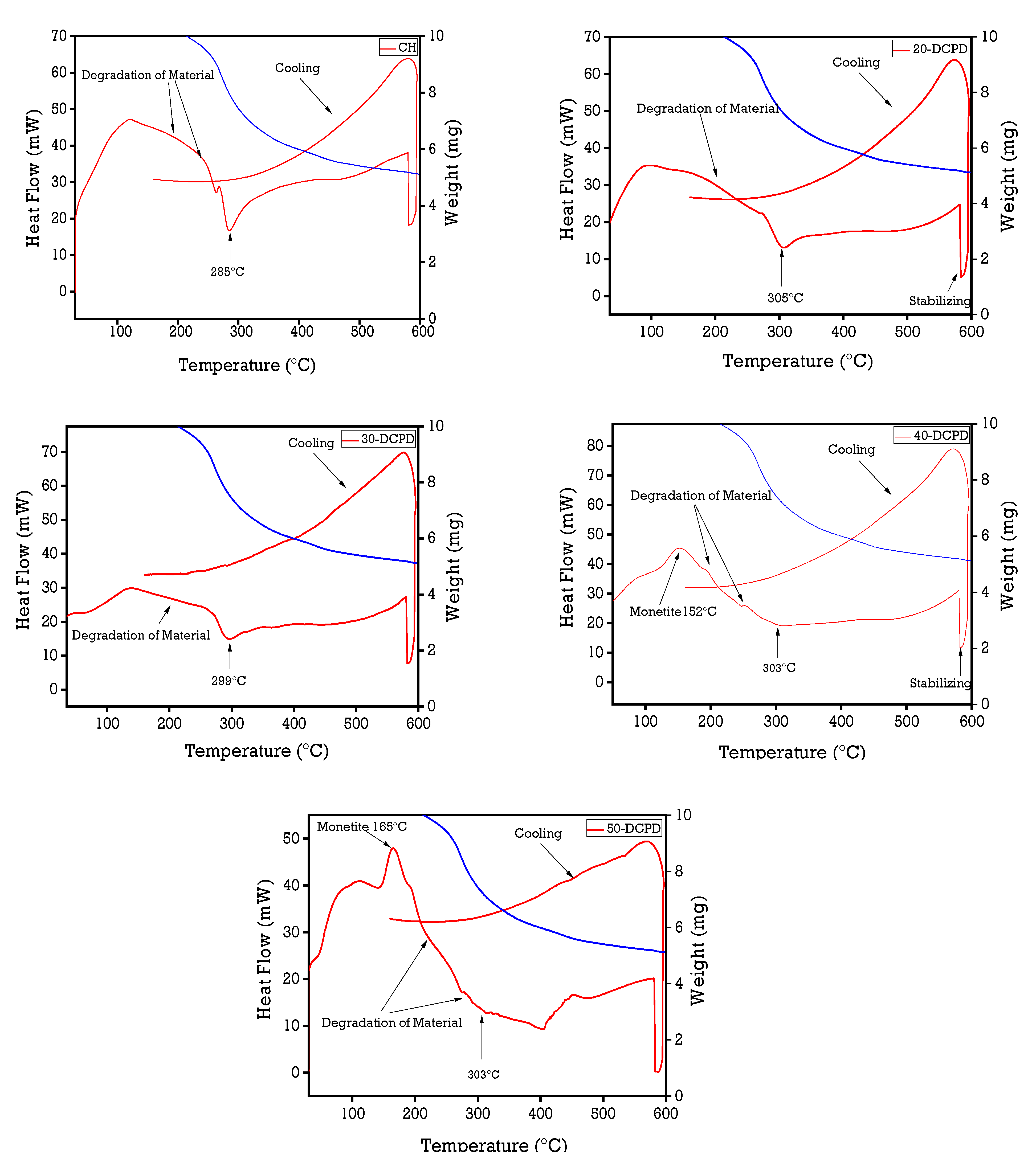
3.4. Simultaneous Thermal Analysis
3.5. Mechanical Properties
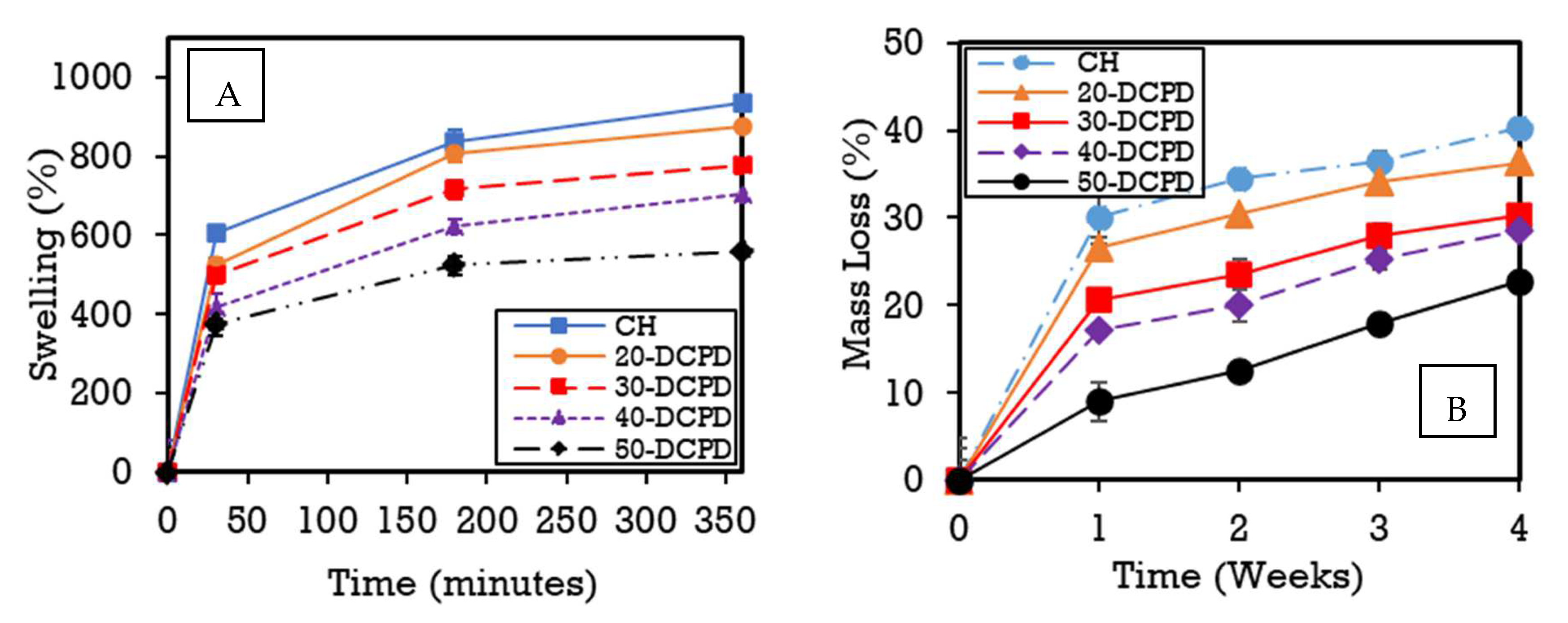
3.6. Scaffold Swelling and Degradation
3.7. Zeta Potential
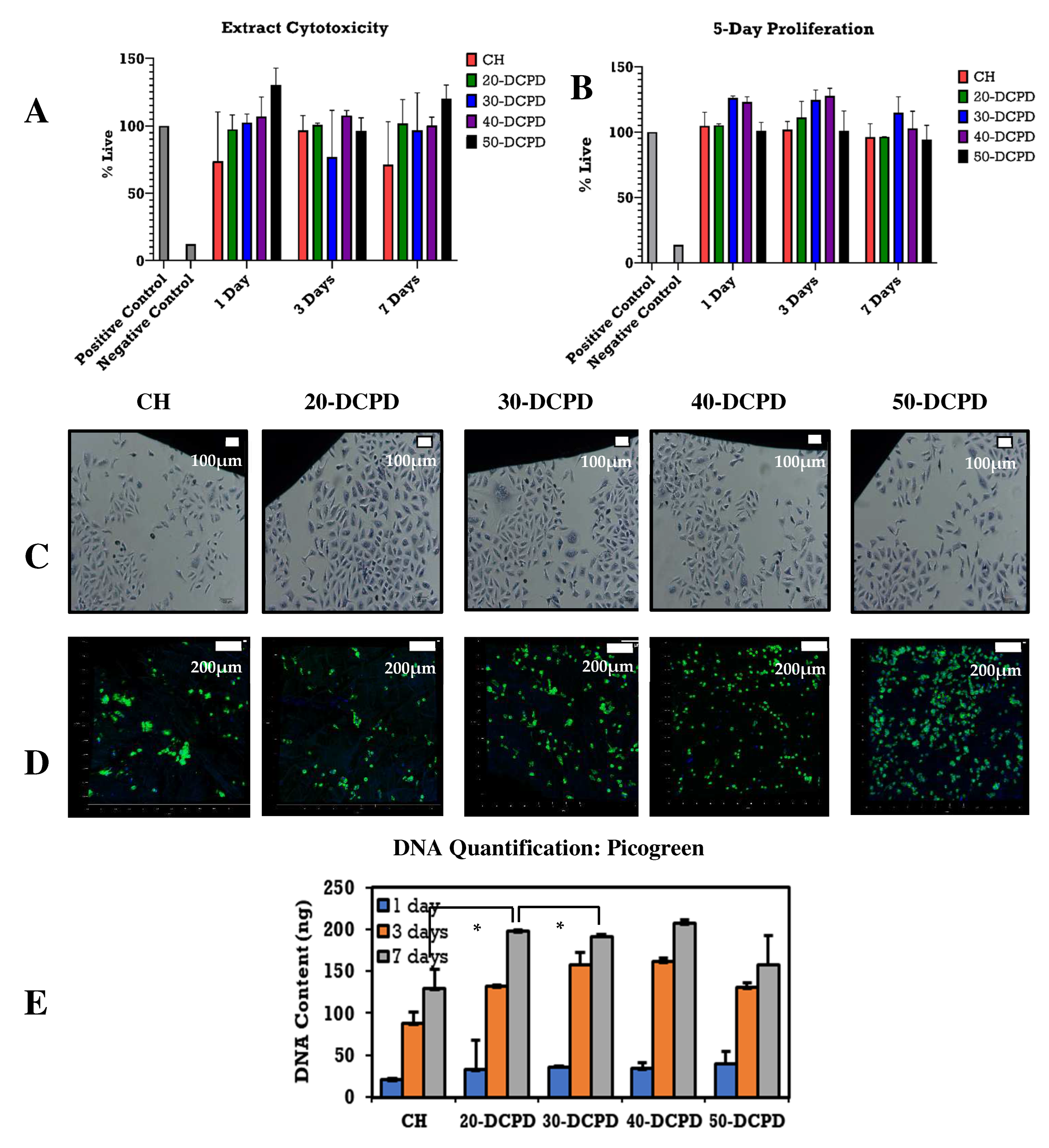
3.8. Extract Cytotoxicity by XTT Assay and DNA Quantification by Picogreen Assay
3.9. Contact Cytotoxicity and Fluorescence Staining
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Levengood, S.K.L.; Zhang, M. Chitosan-based scaffolds for bone tissue engineering. J. Mater. Chem. B 2014, 2, 3161–3184. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Anastasiou, A.; Aslam, Z.; Raif, E.M.; Do, T.; Giannoudis, P.V.; Jha, A. Interrelationships between the structural, spectroscopic, and antibacterial properties of nanoscale (<50 nm) cerium oxides. Sci. Rep. 2021, 11, 20875. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhang, J.; Zhang, C.; Barbieri, D.; Yuan, H.; Moroni, L.; Feng, G. The role of calcium phosphate surface structure in osteogenesis and the mechanisms involved. Acta Biomater. 2020, 106, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, G.; Johnson, B.N.; Jia, X. Three-dimensional (3D) printed scaffold and material selection for bone repair. Acta Biomater. 2019, 84, 16–33. [Google Scholar] [CrossRef]
- Neacsu, I.A.; Serban, A.P.; Nicoara, A.I.; Trusca, R.; Ene, V.L.; Iordache, F. Biomimetic Composite Scaffold Based on Naturally Derived Biomaterials. Polymers 2020, 12, 1161. [Google Scholar] [CrossRef]
- van Vugt, T.A.; Geurts, J.A.P.; Arts, J.J.; Lindfors, N.C. 3—Biomaterials in treatment of orthopedic infections. In Management of Periprosthetic Joint Infections (PJIs); Arts, J.J.C., Geurts, J., Eds.; Woodhead Publishing: Sawston, UK, 2017; pp. 41–68. [Google Scholar] [CrossRef]
- LeGeros, R.Z. Calcium phosphate-based osteoinductive materials. Chem. Rev. 2008, 108, 4742–4753. [Google Scholar] [CrossRef]
- Sheikh, Z.; Abdallah, M.-N.; Hanafi, A.A.; Misbahuddin, S.; Rashid, H.; Glogauer, M. Mechanisms of in Vivo Degradation and Resorption of Calcium Phosphate Based Biomaterials. Materials 2015, 8, 7913–7925. [Google Scholar] [CrossRef]
- Sheikh, Z.; Sima, C.; Glogauer, M. Bone Replacement Materials and Techniques Used for Achieving Vertical Alveolar Bone Augmentation. Materials 2015, 8, 2953–2993. [Google Scholar] [CrossRef]
- Barinov, S.; Komlev, V. Calcium Phosphate Based Bioceramics for Bone Tissue Engineering; Trans Tech Publications: Zurich, Switzerland, 2008. [Google Scholar]
- Lu, J.; Descamps, M.; Dejou, J.; Koubi, G.; Hardouin, P.; Lemaitre, J.; Proust, J.-P. The biodegradation mechanism of calcium phosphate biomaterials in bone. J. Biomed. Mater. Res. 2002, 63, 408–412. [Google Scholar] [CrossRef]
- Barrère, F.; van Blitterswijk, C.A.; de Groot, K. Bone regeneration: Molecular and cellular interactions with calcium phosphate ceramics. Int. J. Nanomed. 2006, 1, 317–332. [Google Scholar]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E. Calcium Phosphates as Delivery Systems for Bisphosphonates. J. Funct. Biomater. 2018, 9, 6. [Google Scholar] [CrossRef]
- Anastasiou, A.D.; Thomson, C.L.; Hussain, S.A.; Edwards, T.J.; Strafford, S.; Malinowski, M.; Mathieson, R.; Brown, C.T.A.; Brown, A.P.; Duggal, M.S.; et al. Sintering of calcium phosphates with a femtosecond pulsed laser for hard tissue engineering. Mater. Des. 2016, 101, 346–354. [Google Scholar] [CrossRef]
- Redepenning, J.; Schlessinger, T.; Burnham, S.; Lippiello, L.; Miyano, J. Characterization of electrolytically prepared brushite and hydroxyapatite coatings on orthopedic alloys. J. Biomed. Mater. Res. 1996, 30, 287–294. [Google Scholar] [CrossRef]
- Fulmer, M.T.; Brown, P.W. Hydrolysis of dicalcium phosphate dihydrate to hydroxyapatite. J. Mater. Sci. Mater. Med. 1998, 9, 197–202. [Google Scholar] [CrossRef]
- Boanini, E.; Gazzano, M.; Bigi, A. Ionic substitutions in calcium phosphates synthesised at low temperature. Acta Biomater. 2010, 6, 1882–1894. [Google Scholar] [CrossRef]
- Laskus, A.; Zgadzaj, A.; Kolmas, J. Selenium-Enriched Brushite: A Novel Biomaterial for Potential Use in Bone Tissue Engineering. Int. J. Mol. Sci. 2018, 19, 4042. [Google Scholar] [CrossRef]
- Rubini, K.; Boanini, E.; Bigi, A. Role of Aspartic and Polyaspartic Acid on the Synthesis and Hydrolysis of Brushite. J. Funct. Biomater. 2019, 10, 11. [Google Scholar] [CrossRef]
- Zhang, Z.-L.; Chen, X.-R.; Bian, S.; Huang, J.; Zhang, T.-L.; Wang, K. Identification of dicalcium phosphate dihydrate deposited during osteoblast mineralisation in vitro. J. Inorg. Biochem. 2014, 131, 109–114. [Google Scholar] [CrossRef]
- Theiss, F.; Apelt, D.; Brand, B.; Kutter, A.; Zlinszky, K.; Bohner, M.; Matter, S.; Frei, C.; Auer, J.A.; von Rechenberg, B. Biocompatibility and resorption of a brushite calcium phosphate cement. Biomaterials 2005, 26, 4383–4394. [Google Scholar] [CrossRef]
- Xia, Z.; Grover, L.M.; Huang, Y.; Adamopoulos, I.E.; Gbureck, U.; Triffitt, J.T.; Shelton, R.M.; Barralet, J.E. In vitro biodegradation of three brushite calcium phosphate cements by a macrophage cell-line. Biomaterials 2006, 27, 4557–4565. [Google Scholar] [CrossRef]
- Tamimi, F.; Kumarasami, B.; Doillon, C.; Gbureck, U.; Le Nihouannen, D.; López-Cabarcos, E.; Barralet, J. Brushite–collagen composites for bone regeneration. Acta Biomater. 2008, 4, 1315–1321. [Google Scholar] [CrossRef]
- Kuemmerle, J.M.; Oberle, A.; Oechslin, C.; Bohner, M.; Frei, C.; Boecken, I.; Rechenberg, B.v. Assessment of the suitability of a new brushite calcium phosphate cement for cranioplasty—An experimental study in sheep. J. Cranio-Maxillofac. Surg. 2005, 33, 37–44. [Google Scholar] [CrossRef]
- Müller, P.; Bulnheim, U.; Diener, A.; Lüthen, F.; Teller, M.; Klinkenberg, E.D.; Neumann, H.G.; Nebe, B.; Liebold, A.; Steinhoff, G.; et al. Calcium phosphate surfaces promote osteogenic differentiation of mesenchymal stem cells. J. Cell. Mol. Med. 2008, 12, 281–291. [Google Scholar] [CrossRef]
- Yuan, H.; Fernandes, H.; Habibovic, P.; de Boer, J.; Barradas, A.M.C.; de Ruiter, A.; Walsh, W.R.; van Blitterswijk, C.A.; de Bruijn, J.D. Osteoinductive ceramics as a synthetic alternative to autologous bone grafting. Proc. Natl. Acad. Sci. USA 2010, 107, 13614–13619. [Google Scholar] [CrossRef]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Roberts, G.A.F. Structure of Chitin and Chitosan. In Chitin Chemistry; Roberts, G.A.F., Ed.; Macmillan Education: London, UK, 1992; pp. 1–53. [Google Scholar] [CrossRef]
- Aranaz, I.; Martínez-Campos, E.; Moreno-Vicente, C.; Civantos, A.; García-Arguelles, S.; Del Monte, F. Macroporous Calcium Phosphate/Chitosan Composites Prepared via Unidirectional Ice Segregation and Subsequent Freeze-Drying. Materials 2017, 10, 516. [Google Scholar] [CrossRef]
- Anitha, A.; Sowmya, S.; Kumar, P.T.S.; Deepthi, S.; Chennazhi, K.P.; Ehrlich, H.; Tsurkan, M.; Jayakumar, R. Chitin and chitosan in selected biomedical applications. Prog. Polym. Sci. 2014, 39, 1644–1667. [Google Scholar] [CrossRef]
- Di Martino, A.; Sittinger, M.; Risbud, M.V. Chitosan: A versatile biopolymer for orthopaedic tissue-engineering. Biomaterials 2005, 26, 5983–5990. [Google Scholar] [CrossRef]
- Aljawish, A.; Chevalot, I.; Jasniewski, J.; Scher, J.; Muniglia, L. Enzymatic synthesis of chitosan derivatives and their potential applications. J. Mol. Catal. B Enzym. 2015, 112, 25–39. [Google Scholar] [CrossRef]
- Zhang, M.; Li, X.H.; Gong, Y.D.; Zhao, N.M.; Zhang, X.F. Properties and biocompatibility of chitosan films modified by blending with PEG. Biomaterials 2002, 23, 2641–2648. [Google Scholar] [CrossRef]
- Shanmugasundaram, N.; Ravichandran, P.; Reddy, P.N.; Ramamurty, N.; Pal, S.; Rao, K.P. Collagen-chitosan polymeric scaffolds for the in vitro culture of human epidermoid carcinoma cells. Biomaterials 2001, 22, 1943–1951. [Google Scholar] [CrossRef]
- Lahiji, A.; Sohrabi, A.; Hungerford, D.S.; Frondoza, C.G. Chitosan supports the expression of extracellular matrix proteins in human osteoblasts and chondrocytes. J. Biomed. Mater. Res. 2000, 51, 586–595. [Google Scholar] [CrossRef]
- Mi, F.-L.; Shyu, S.-S.; Wu, Y.-B.; Lee, S.-T.; Shyong, J.-Y.; Huang, R.-N. Fabrication and characterisation of a sponge-like asymmetric chitosan membrane as a wound dressing. Biomaterials 2001, 22, 165–173. [Google Scholar] [CrossRef]
- Younes, I.; Rinaudo, M. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Rezwan, K.; Chen, Q.Z.; Blaker, J.J.; Boccaccini, A.R. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef]
- Rodríguez-Vázquez, M.; Vega-Ruiz, B.; Ramos-Zúñiga, R.; Saldaña-Koppel, D.A.; Quiñones-Olvera, L.F. Chitosan and Its Potential Use as a Scaffold for Tissue Engineering in Regenerative Medicine. BioMed Res. Int. 2015, 2015, 821279. [Google Scholar] [CrossRef] [PubMed]
- Croisier, F.; Jérôme, C. Chitosan-based biomaterials for tissue engineering. Eur. Polym. J. 2013, 49, 780–792. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Mohamed, K.R.; El-Rashidy, Z.M.; Salama, A.A. In vitro properties of nano-hydroxyapatite/chitosan biocomposites. Ceram. Int. 2011, 37, 3265–3271. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, G.; Wu, Q.; Zuo, J.; Qin, Y.; Wang, J. Novel Mesoporous Hydroxyapatite/Chitosan Composite for Bone Repair. J. Bionic Eng. 2012, 9, 243–251. [Google Scholar] [CrossRef]
- Lee, J.S.; Baek, S.D.; Venkatesan, J.; Bhatnagar, I.; Chang, H.K.; Kim, H.T.; Kim, S.-K. In vivo study of chitosan-natural nano hydroxyapatite scaffolds for bone tissue regeneration. Int. J. Biol. Macromol. 2014, 67, 360–366. [Google Scholar] [CrossRef]
- Seol, Y.-J.; Lee, J.-Y.; Park, Y.-J.; Lee, Y.-M.; Ku, Y.; Rhyu, I.-C.; Lee, S.-J.; Han, S.-B.; Chung, C.-P. Chitosan sponges as tissue engineering scaffolds for bone formation. Biotechnol. Lett. 2004, 26, 1037–1041. [Google Scholar] [CrossRef]
- Sultankulov, B.; Berillo, D.; Sultankulova, K.; Tokay, T.; Saparov, A. Progress in the Development of Chitosan-Based Biomaterials for Tissue Engineering and Regenerative Medicine. Biomolecules 2019, 9, 470. [Google Scholar] [CrossRef]
- Kiuchi, H.; Kai, W.; Inoue, Y. Preparation and characterisation of poly(ethylene glycol) crosslinked chitosan films. J. Appl. Polym. Sci. 2008, 107, 3823–3830. [Google Scholar] [CrossRef]
- Adekogbe, I.; Ghanem, A. Fabrication and characterisation of DTBP-crosslinked chitosan scaffolds for skin tissue engineering. Biomaterials 2005, 26, 7241–7250. [Google Scholar] [CrossRef]
- Yin, Y.; Ye, F.; Cui, J.; Zhang, F.; Li, X.; Yao, K. Preparation and characterisation of macroporous chitosan-gelatin/beta-tricalcium phosphate composite scaffolds for bone tissue engineering. J. Biomed. Mater. Res. A 2003, 67, 844–855. [Google Scholar] [CrossRef]
- Yasmeen, S.; Kabiraz, M.; Saha, B.; Qadir, M.; Gafur, M.; Masum, S. Chromium (VI) Ions Removal from Tannery Effluent using Chitosan-Microcrystalline Cellulose Composite as Adsorbent. Int. Res. J. Pure Appl. Chem. 2016, 10, 23315. [Google Scholar] [CrossRef]
- Mandel, S.; Tas, A.C. Brushite (CaHPO4·2H2O) to octacalcium phosphate (Ca8(HPO4)2(PO4)4·5H2O) transformation in DMEM solutions at 36.5 °C. Mater. Sci. Eng. C Mater. Biol. Appl. 2010, 30, 245–254. [Google Scholar] [CrossRef]
- Lee, D.; Kumta, P.N. Chemical synthesis and stabilisation of magnesium substituted brushite. Mater. Sci. Eng. C 2010, 30, 934–943. [Google Scholar] [CrossRef]
- Singh, S.; Singh, V.; Aggarwal, S.; Mandal, U. Synthesis of brushite nanoparticles at different temperatures. Chem. Pap. 2010, 64, 491. [Google Scholar] [CrossRef]
- Mai, T.; Wolski, K.; Puciul-Malinowska, A.; Kopyshev, A.; Gräf, R.; Bruns, M.; Zapotoczny, S.; Taubert, A. Anionic Polymer Brushes for Biomimetic Calcium Phosphate Mineralization-A Surface with Application Potential in Biomaterials. Polymers 2018, 10, 1165. [Google Scholar] [CrossRef] [PubMed]
- Fadeeva, I.V.; Barinov, S.M.; Fedotov, A.Y.; Komlev, V.S. Interactions of calcium phosphates with chitosan. Dokl. Chem. 2011, 441, 387–390. [Google Scholar] [CrossRef]
- Wan, Y.; Creber, K.A.M.; Peppley, B.; Bui, V.T. Synthesis, Characterisation and Ionic Conductive Properties of Phosphorylated Chitosan Membranes. Macromol. Chem. Phys. 2003, 204, 850–858. [Google Scholar] [CrossRef]
- Dantas, M.J.L.; dos Santos, B.F.F.; Tavares, A.A.; Maciel, M.A.; Lucena, B.M.; Fook, M.V.L.; Silva, S.M.d.L. The Impact of the Ionic Cross-Linking Mode on the Physical and In Vitro Dexamethasone Release Properties of Chitosan/Hydroxyapatite Beads. Molecules 2019, 24, 4510. [Google Scholar] [CrossRef]
- Gritsch, L.; Maqbool, M.; Mouriño, V.; Ciraldo, F.E.; Cresswell, M.; Jackson, P.R.; Lovell, C.; Boccaccini, A.R. Chitosan/hydroxyapatite composite bone tissue engineering scaffolds with dual and decoupled therapeutic ion delivery: Copper and strontium. J. Mater. Chem. B 2019, 7, 6109–6124. [Google Scholar] [CrossRef]
- Leceta, I.; Guerrero, P.; Ibarburu, I.; Duenas, M.T.; de la Caba, K. Characterization and antimicrobial analysis of chitosan-based films. J. Food Eng. 2013, 116, 889–899. [Google Scholar] [CrossRef]
- Lewandowska, K. Miscibility and thermal stability of poly(vinyl alcohol)/chitosan mixtures. Thermochim. Acta 2009, 493, 42–48. [Google Scholar] [CrossRef]
- Ruini, F.; Tonda-Turo, C.; Chiono, V.; Ciardelli, G. Chitosan membranes for tissue engineering: Comparison of different crosslinkers. Biomed. Mater. 2015, 10, 065002. [Google Scholar] [CrossRef]
- Mucha, M.; Pawlak, A. Complex study on chitosan degradability. Polimery 2002, 47, 509–516. [Google Scholar] [CrossRef]
- Szymańska, E.; Winnicka, K. Stability of chitosan-a challenge for pharmaceutical and biomedical applications. Mar. Drugs 2015, 13, 1819–1846. [Google Scholar] [CrossRef]
- Adair, J.H.; Suvaci, E.; Sindel, J. Surface and Colloid Chemistry. In Encyclopedia of Materials: Science and Technology; Buschow, K.H.J., Cahn, R.W., Flemings, M.C., Ilschner, B., Kramer, E.J., Mahajan, S., Veyssière, P., Eds.; Elsevier: Oxford, UK, 2001; pp. 1–10. [Google Scholar] [CrossRef]
- Asghar, W.; Islam, M.; Wadajkar, A.; Wan, Y.; Ilyas, A.; Nguyen, K.; Iqbal, S. PLGA Micro and Nanoparticles Loaded into Gelatin Scaffold for Controlled Drug Release. IEEE Trans. Nanotechnol. 2012, 11, 546–553. [Google Scholar] [CrossRef]
- Couvreur, P.; Barratt, G.; Fattal, E.; Legrand, P.; Vauthier, C. Nanocapsule technology: A review. Crit. Rev. Ther. Drug Carr. Syst. 2002, 19, 99–134. [Google Scholar] [CrossRef]
- Madihally, S.V.; Matthew, H.W. Porous chitosan scaffolds for tissue engineering. Biomaterials 1999, 20, 1133–1142. [Google Scholar] [CrossRef]
- Reys, L.L.; Silva, S.S.; Oliveira, J.M.; Caridade, S.G.; Mano, J.F.; Silva, T.H.; Reis, R.L. Revealing the potential of squid chitosan-based structures for biomedical applications. Biomed. Mater. 2013, 8, 045002. [Google Scholar] [CrossRef]
- Wang, W.; Yeung, K.W.K. Bone grafts and biomaterials substitutes for bone defect repair: A review. Bioact. Mater. 2017, 2, 224–247. [Google Scholar] [CrossRef]
- Le Huec, J.C.; Schaeverbeke, T.; Clement, D.; Faber, J.; Le Rebeller, A. Influence of porosity on the mechanical resistance of hydroxyapatite ceramics under compressive stress. Biomaterials 1995, 16, 113–118. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.W.; Lee, Y.M.; Lee, H.B.; Khang, G. Macroporous biodegradable natural/synthetic hybrid scaffolds as small intestine submucosa impregnated poly(D,L-lactide-co-glycolide) for tissue-engineered bone. J. Biomater. Sci. Polym. Ed. 2004, 15, 1003–1017. [Google Scholar] [CrossRef]
- Yu, J.; Xia, H.; Ni, Q.-Q. A three-dimensional porous hydroxyapatite nanocomposite scaffold with shape memory effect for bone tissue engineering. J. Mater. Sci. 2017, 53, 4734–4744. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Rodrigues, M.T.; Silva, S.S.; Malafaya, P.B.; Gomes, M.E.; Viegas, C.A.; Dias, I.R.; Azevedo, J.T.; Mano, J.F.; Reis, R.L. Novel hydroxyapatite/chitosan bilayered scaffold for osteochondral tissue-engineering applications: Scaffold design and its performance when seeded with goat bone marrow stromal cells. Biomaterials 2006, 27, 6123–6137. [Google Scholar] [CrossRef]
- Klawitter, J.J.; Bagwell, J.G.; Weinstein, A.M.; Sauer, B.W.; Pruitt, J.R. An evaluation of bone growth into porous high density polyethylene. J. Biomed. Mater. Res. 1976, 10, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Kramschuster, A.; Turng, L.-S. 17—Fabrication of Tissue Engineering Scaffolds. In Handbook of Biopolymers and Biodegradable Plastics; Ebnesajjad, S., Ed.; William Andrew Publishing: Boston, MA, USA, 2013; pp. 427–446. [Google Scholar] [CrossRef]
- Yannas, I.V. Tissue regeneration by use of collagen-glycosaminoglycan copolymers. Clin. Mater. 1992, 9, 179–187. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef] [PubMed]
- Velasco, M.A.; Narváez-Tovar, C.A.; Garzón-Alvarado, D.A. Design, Materials, and Mechanobiology of Biodegradable Scaffolds for Bone Tissue Engineering. BioMed Res. Int. 2015, 2015, 729076. [Google Scholar] [CrossRef]
- Chocholata, P.; Kulda, V.; Babuska, V. Fabrication of Scaffolds for Bone-Tissue Regeneration. Materials 2019, 12, 568. [Google Scholar] [CrossRef]
- Yang, L.; Meiwen, A.; Hai-Xia, Q.; Li, W. The Properties of Chitosan-Gelatin Scaffolds by Once or Twice Vacuum Freeze-Drying Methods. Polym.-Plast. Technol. Eng. 2013, 52, 1154–1159. [Google Scholar] [CrossRef]
- Peter, M.; Ganesh, N.; Selvamurugan, N.; Nair, S.V.; Furuike, T.; Tamura, H.; Jayakumar, R. Preparation and characterisation of chitosan–gelatin/nanohydroxyapatite composite scaffolds for tissue engineering applications. Carbohydr. Polym. 2010, 80, 687–694. [Google Scholar] [CrossRef]
- Mallika, P.; Himabindu, A.; Shailaja, D. Modification of chitosan towards a biomaterial with improved physico-chemical properties. J. Appl. Polym. Sci. 2006, 101, 63–69. [Google Scholar] [CrossRef]
- Liuyun, J.; Yubao, L.; Chengdong, X. A novel composite membrane of chitosan-carboxymethyl cellulose polyelectrolyte complex membrane filled with nano-hydroxyapatite I. Preparation and properties. J. Mater. Sci. Mater. Med. 2009, 20, 1645–1652. [Google Scholar] [CrossRef]
- Farag, R.K.; Mohamed, R.R. Synthesis and characterisation of carboxymethyl chitosan nanogels for swelling studies and antimicrobial activity. Molecules 2012, 18, 190–203. [Google Scholar] [CrossRef]
- Ahmadi, F.; Oveisi, Z.; Samani, S.M.; Amoozgar, Z. Chitosan based hydrogels: Characteristics and pharmaceutical applications. Res. Pharm. Sci. 2015, 10, 1–16. [Google Scholar]
- Kim, S.J.; Shin, S.R.; Lee, Y.M.; Kim, S.I. Swelling characterisations of chitosan and polyacrylonitrile semi-interpenetrating polymer network hydrogels. J. Appl. Polym. Sci. 2003, 87, 2011–2015. [Google Scholar] [CrossRef]
- Costa, E.S., Jr.; Barbosa-Stancioli, E.F.; Mansur, A.A.P.; Vasconcelos, W.L.; Mansur, H.S. Preparation and characterisation of chitosan/poly(vinyl alcohol) chemically crosslinked blends for biomedical applications. Carbohydr. Polym. 2009, 76, 472–481. [Google Scholar] [CrossRef]
- Forman, S.; Káš, J.; Fini, F.; Steinberg, M.; Ruml, T. The effect of different solvents on the ATP/ADP content and growth properties of HeLa cells. J. Biochem. Mol. Toxicol. 1999, 13, 11–15. [Google Scholar] [CrossRef]
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef]
- Islam, N.; Dmour, I.; Taha, M.O. Degradability of chitosan micro/nanoparticles for pulmonary drug delivery. Heliyon 2019, 5, e01684. [Google Scholar] [CrossRef]
- Ing, L.Y.; Zin, N.M.; Sarwar, A.; Katas, H. Antifungal Activity of Chitosan Nanoparticles and Correlation with Their Physical Properties. Int. J. Biomater. 2012, 2012, 632698. [Google Scholar] [CrossRef]
- Katas, H.; Alpar, H.O. Development and characterisation of chitosan nanoparticles for siRNA delivery. J. Control. Release 2006, 115, 216–225. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Description | Chemical Formula | CH:DCPD |
|---|---|---|---|
| DCPD | Dicalcium Phosphate Dihydrate | CaHPO4·2H2O | 0:100 |
| CH | Chitosan scaffold | (C6H11NO4)n | 100:0 |
| 20-DCPD | Mineral loaded scaffold | - | 80:20 |
| 30-DCPD | Mineral loaded scaffold | - | 70:30 |
| 40-DCPD | Mineral loaded scaffold | - | 60:40 |
| 50-DCPD | Mineral loaded scaffold | - | 50:50 |
| Sample | Diffraction Plane (h k l) | Crystallite Size (nm) | Crystallinity (%) | |
|---|---|---|---|---|
| (0 2 0) | (0 4 0) | |||
| CH | - | - | 0.9059 | 0.1104 |
| 20-DCPD | 11.53° | 20.64° | 2.2404 | 20.6399 |
| 30-DCPD | 11.93° | 21.04° | 11.2882 | 29.3773 |
| 40-DCPD | 11.53° | 20.64° | 25.9477 | 38.7656 |
| 50-DCPD | 11.14° | 20.44° | 26.4008 | 69.8635 |
| DCPD | 10.97° | 22.75° | 41.2679 | 78.5123 |
| CH | 20-DCPD | 30-DCPD | 40-DCPD | 50-DCPD | |
|---|---|---|---|---|---|
| Young’s Modulus (kN/m2) | 5.38 ± 0.03 | 7.26 ± 0.06 | 20.92 ± 0.11 | 22.73 ± 2.10 | 25.50 ± 0.54 |
| Tensile Strength (kPa) | 7.07 ± 0.03 | 9.86 ± 0.05 | 26.84 ± 0.08 | 23.21 ± 0.63 | 27.13 ± 0.25 |
| Sample | Zeta Potential (mV) | Standard Deviation |
|---|---|---|
| DCPD | −12.44 | 0.41 |
| CH | +43.47 | 0.35 |
| 20-DCPD | +39.57 | 0.31 |
| 30-DCPD | +34.53 | 0.38 |
| 40-DCPD | +28.23 | 0.25 |
| 50-DCPD | +20.23 | 0.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, N.; Braxton, T.M.; Anastasiou, A.; Raif, E.M.; Chung, C.K.Y.; Kumar, S.; Giannoudis, P.V.; Jha, A. Dicalcium Phosphate Dihydrate Mineral Loaded Freeze-Dried Scaffolds for Potential Synthetic Bone Applications. Materials 2022, 15, 6245. https://doi.org/10.3390/ma15186245
Iqbal N, Braxton TM, Anastasiou A, Raif EM, Chung CKY, Kumar S, Giannoudis PV, Jha A. Dicalcium Phosphate Dihydrate Mineral Loaded Freeze-Dried Scaffolds for Potential Synthetic Bone Applications. Materials. 2022; 15(18):6245. https://doi.org/10.3390/ma15186245
Chicago/Turabian StyleIqbal, Neelam, Thomas Michael Braxton, Antonios Anastasiou, El Mostafa Raif, Charles Kai Yin Chung, Sandeep Kumar, Peter V. Giannoudis, and Animesh Jha. 2022. "Dicalcium Phosphate Dihydrate Mineral Loaded Freeze-Dried Scaffolds for Potential Synthetic Bone Applications" Materials 15, no. 18: 6245. https://doi.org/10.3390/ma15186245
APA StyleIqbal, N., Braxton, T. M., Anastasiou, A., Raif, E. M., Chung, C. K. Y., Kumar, S., Giannoudis, P. V., & Jha, A. (2022). Dicalcium Phosphate Dihydrate Mineral Loaded Freeze-Dried Scaffolds for Potential Synthetic Bone Applications. Materials, 15(18), 6245. https://doi.org/10.3390/ma15186245