1. Introduction
Currently, the problem of the depletion of fossil natural resources with the accompanying complication of the global environmental situation is closely related to the increase in the demand for energy sources, fuels and chemicals. Therefore, the opportunity to use renewable raw materials, in particular biomass and biodiesel, to solve the challenges of modern energy as well as the development of new methods for biomass conversion into valuable organic intermediates used in various industries are among the trending research areas. Biomass is a renewable source of raw materials to produce fuels and a number of important organic compounds for chemical industries. Thus, polyatomic alcohols, in particular propylene glycol (PG) and glycerol, are the products of biomass conversion [
1,
2,
3]. It is noteworthy that PG can also be obtained by the glycerol hydrogenolysis [
4].
Experimental studies of the PG selective oxidation into carbonyl/carboxylic compounds (methylglyoxal, hydroxyacetone, etc.) are known in the literature for catalysts with supported metal nanoparticles, including Ag [
5,
6,
7], Pt [
8] and Pd [
9,
10,
11,
12]. The mechanism of PG conversion was theoretically studied over the Ag
4 cluster [
13]. As in the case of ethylene glycol oxidation [
9,
14,
15], the PG adsorption on the surface of single crystal Ag (110) and Pd (111) coated with oxygen led to the breaking of O–H bonds with the formation of the intermediate 1,2-propanedioxy (–OCH(CH
3)CH
2O–, PDO) [
5,
6,
9]. As the temperature increased, the PDO transformations yielded a number of products, including hydroxyacetone or acetol (CH
3COCH
2OH), lactaldehyde (CH
3CH(OH)CHO), and methylglyoxal or pyruvaldehyde (CH
3COCHO). Moreover, methylglyoxal and hydroxyacetone are of particular interest as the key intermediates with unique chemical properties due to the presence of two oxygen-containing groups. In Refs. [
5,
6], it was shown that, first of all, the C–H bond breaking on Ag (110) occurred at the secondary carbon atom of the PDO intermediate as evidenced by the low temperature of hydroxyacetone desorption. In Ref. [
9], using the adsorption of ethylene glycol and PG on Pd (111) as an example it was shown that the intermediate compounds were strongly bound to the surface with Pd (111) in a planar orientation; as a result, glyoxal and methylglyoxal were desorbed in small amounts at temperatures above 250 K, however, they were primarily decarbonylated to form CO, H
2 and methane. In Ref. [
16], it was suggested that in the case of using the metal/bimetallic catalysts in the oxidation of polyatomic alcohols (e.g., PG and glycerol), the key role of the modifying metal was connected with controlling the reagent bond strength with the catalyst surface without a sharp change in the preferred geometry of the adsorbate, and such changes of binding energies can have a strong impact on the catalytic properties. Transition metals, such as Bi for Pd-Bi supported catalysts [
12], are often used as a modifying metal added to those of the Pt or Au subgroups.
Catalysts based on transition metal compounds, such as Mo, Fe, V, are catalytically active in various oxidation processes making it possible to consider them as a promising alternative to those based on noble metals for selective conversion of alcohols and methane. In Ref. [
17], the ethylene glycol reactions on the surface of the Mo (110) single crystal were considered in details by a complex of physical-chemical methods. It was shown that during the ethylene glycol adsorption on the surface of Mo (110), the intermediate compounds with the Mo-O bond were formed both in mono- (–OCH
2CH
2OH) and bidentate (–OCH
2CH
2O–) configurations. The authors found that bidentate species were more reactive due to their stability and greater propensity to form ethylene as compared to the potential transformations of the monodentate intermediates. High strength of the O–Mo bond caused the ethylene glycol deoxygenation to yield ethylene.
In Ref. [
18], FePO
4 catalysts were studied in direct oxidation of methane to methanol. Using Mössbauer spectroscopy coupled with XRD, the authors studied in details the phase transformations of the FePO
4 catalysts in atmospheres of various oxidizers O
2, H
2O, and N
2O. It was shown that the Fe
2P
2O
7 phase dominated in the reduced catalyst. The use of H
2O and N
2O as oxidants in the selective conversion of methane to methanol promoted the formation of an active and selective mixed phase α-Fe
3(P
2O
7)
2 (Fe
3+, Fe
2+), while the formation of the less active phase β-Fe
3(P
2O
7)
2 was observed to a lesser extent, and the amount of the Fe
2P
2O
7 phase decreased accordingly. It is worth noting that the Fe valence states can strongly affect the performance of Fe-based catalysts [
19,
20].
Due to the important industrial applications, the study of the surfaces of oxide iron-molybdenum catalysts for selective methanol oxidation into formaldehyde continues to attract the research attention [
21,
22]. In Ref. [
22], the influence of the preparation method on the stability of the synthesized and industrial iron–molybdenum catalysts were studied. Thus, the catalysts prepared by hydrothermal method contained an excess of molybdenum represented by the metastable h-MoO
3 phase, which, upon calcination, transformed into the thermodynamically stable α-MoO
3 phase. The authors found that the crystal structure, crystal size and/or morphology of MoO
3, taken in excess with respect to the iron molybdate, had a significant effect on the stability of the iron-molybdenum catalysts.
An approach involving the silver incorporation into the complex framework zirconium phosphates to prepare the catalysts active in the selective oxidation/dehydrogenation of ethanol to acetaldehyde was proposed in Ref. [
23]. It was shown that the silver addition to complex zirconium phosphate catalysts led to a selectivity increase in the ethanol oxidation to acetaldehyde up to 74% at 330 °C at 93% ethanol conversion, significantly reducing the conversion along the dehydration route. The combination of redox and acid-base sites on the catalyst surface made it possible to control the main directions of transformations of polyatomic alcohols.
Thus, the studies aimed at finding the approaches and methods to assess the reactivity of different catalysts based on transition metal compounds, in particular Fe, using quantum-chemical calculations, are currently relevant to predict the surface properties of multicomponent catalysts. In previous works, when studying a series of supported FePO
4/SiO
2 [
24] and Fe-Mo-O/SiO
2 catalysts [
25,
26], it was shown that such catalysts were active and selective in the reaction of vapor-phase PG oxidation to methylglyoxal.
The present work is devoted to a comparison of the catalytic properties of FePO4/SiO2 and Fe-Mo-O/SiO2 catalysts using theoretical and experimental approaches. It has been shown that the theoretical estimation of the substrate–catalyst binding energy in comparison with the catalytic data can be a key to estimate the reactivity of intermediates depending on the chemical surrounding of the Fe sites.
3. Results and Discussion
Table 1 lists the designations of the synthesized catalysts, the amounts of deposited components according to the XRF results and the specific surface area. Both iron-containing catalysts are characterized by similar Fe content (per iron (III) oxide) as well as a close molar ratio [P or Mo]/Fe.
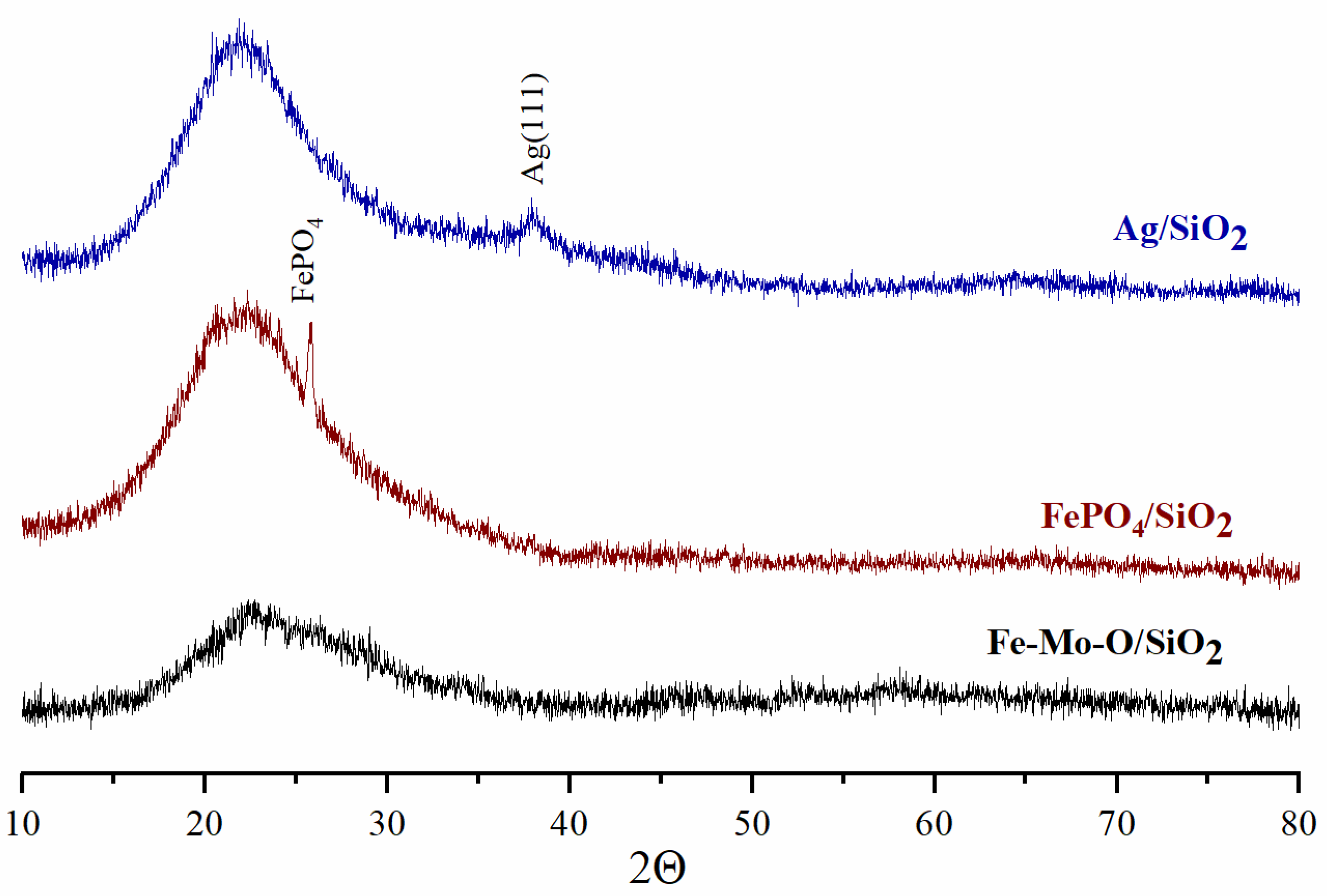
Figure 1 shows the XRD patterns for the supported catalysts and reference samples. The appearance of a wide halo in the range of 2Θ angles up to 30° associated with the amorphous state of SiO
2 is observed in the XRD patterns for all deposited samples.
Figure 1 shows that no crystalline phases are observed by XRD in the Fe-Mo-O/SiO
2 sample. The XRD pattern of the FePO
4/SiO
2 sample features a reflection that corresponds to the FePO
4 iron orthophosphate phase (No. 01-077-0094). The low-intensity reflection in the range of 38° 2Θ in the XRD pattern for the Ag/SiO
2 reference sample corresponds to the metallic Ag(111) phase.
The catalytic properties of the prepared catalysts FePO
4/SiO
2, Fe-Mo-O/SiO
2 and Ag/SiO
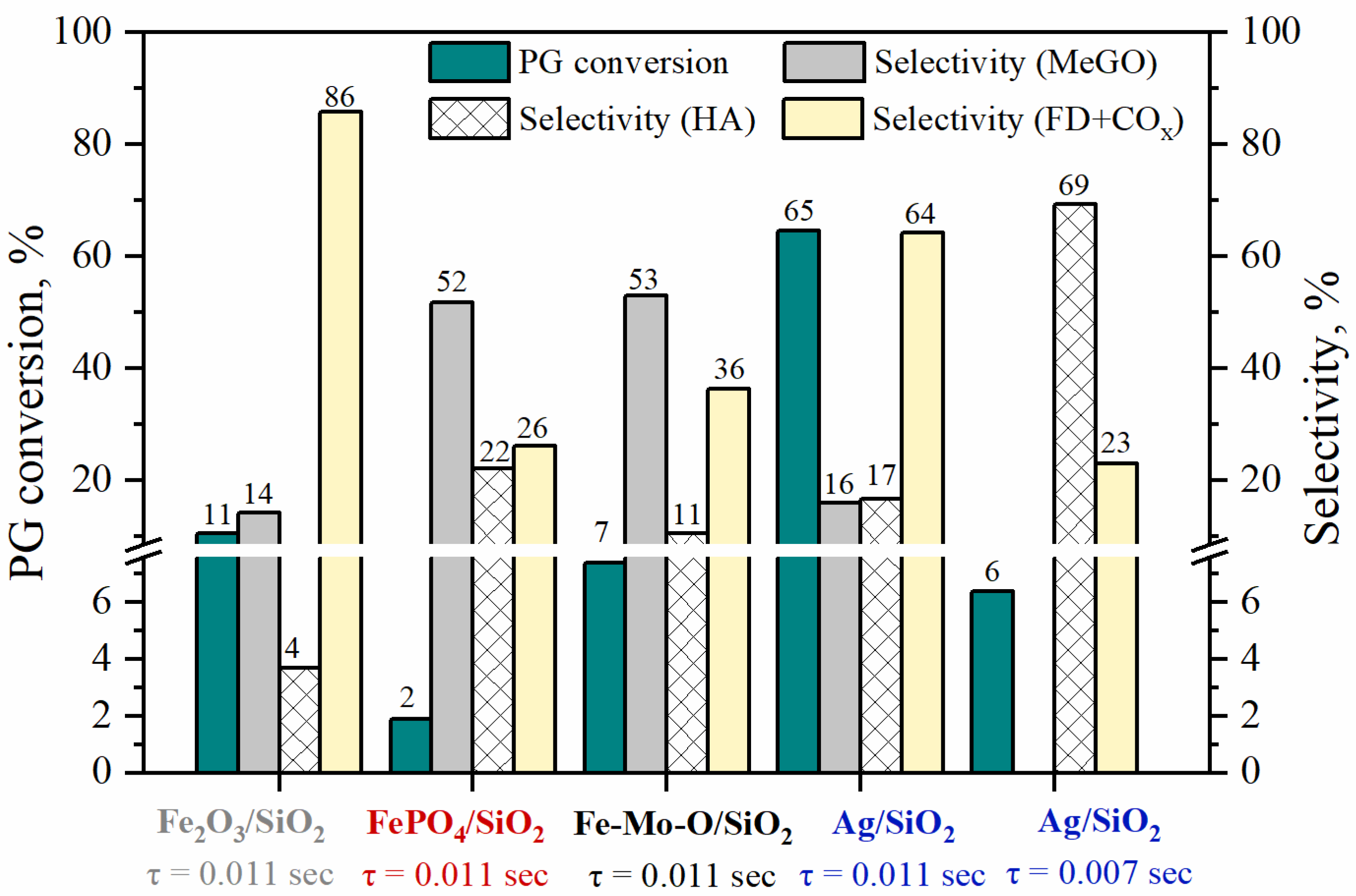
2 (reference sample) were studied in the reaction of vapor-phase PG oxidation to methylglyoxal under the same conditions for all catalysts: contact time was 0.011 s (m
cat = 0.15 g) and temperature was 350 °C. For the Ag/SiO
2 sample, the additional catalytic experiments were carried out at a decreased contact time of 0.007 s (m
cat = 0.1 g).
Figure 2 represents the results obtained.
Thus, in the case of iron-containing catalysts, at the PG conversion of 2 to 7%, the total selectivity towards C
3 products is ~70%, including ~50% towards methylglyoxal for both FePO
4/SiO
2 and Fe-Mo-O/SiO
2 catalysts. The contribution of the side processes with the cleavage of the C–C bond yielding C
1 products (formaldehyde, CO
x) on these catalysts is 26% and 36%, respectively. A Fe
2O
3/SiO
2 reference sample shows higher activity in PG oxidation when compared with samples FePO
4/SiO
2 and Fe-Mo-O/SiO
2. However, in the case of pristine Fe
2O
3/SiO
2 catalyst, the C–C bond cleavage route is the dominant one, and the total selectivity of formation of the C–C bond cleavage products reaches 86% (
Figure 2), while the selectivity towards the desired methylglyoxal does not exceed 20%. The use of FePO
4/SiO
2 and Fe-Mo-O/SiO
2 catalysts permits to achieve high selectivity towards methylglyoxal while keeping the PG conversion in the case of Mo introduction in the composition of Fe-containing catalyst.
For the Ag-containing catalyst at a contact time of 0.011 s, an increase in the PG conversion to up to 65% is accompanied by a decrease in the total selectivity towards C
3 products up to 33% (selectivities towards MeGO and HA are 16% and 17%). For correct comparison of the catalytic properties of the Fe-containing catalysts with Ag/SiO
2, the additional experiments are carried out for the Ag-containing catalyst with a contact time reduced to 0.007 s. At low PG conversion (6%), hydroxyacetone is the main reaction product with a selectivity of ~70%, and methylglyoxal is not formed at this contact time (
Figure 2). The formation of products of C–C bond cleavage under these conditions is reduced to up to 23% that is comparable to iron-containing catalysts.
The iron phosphate catalyst exhibits the lowest activity in PG selective oxidation, while the selectivity towards the target product methylglyoxal reaches 52%, and the selectivity towards by-products does not exceed 30%. For the iron-molybdenum catalyst, a noticeable increase in the PG conversion (up to 7%) is observed, while maintaining high selectivity towards methylglyoxal (
Figure 2). Such an effect can be associated with the energies of interaction of the reagents with the surface-active sites of the supported FePO
4 and Fe
2(MoO
4)
3 catalysts and the structure of the formed intermediate compounds.
Maintaining high selectivity towards C
3 products (hydroxyacetone) for the silver-containing supported catalyst is possible only if the contact time is reduced by almost a factor of 2 (from 0.011 to 0.007 s). At low contact time and 350 °C, the PG adsorption on the surface of the Ag/SiO
2 catalyst occurs predominantly in the monodentate configuration with the participation of OH groups on the silica surface [
15].
Thus, at low PG conversion (<10%), the iron-containing catalysts are characterized by high selectivity towards C3 products, methylglyoxal (50–53%) and hydroxyacetone (up to 22%), while high selectivity is observed on the Ag-containing catalyst only towards hydroxyacetone (~70%), methylglyoxal is not detected in the product composition. Thus, in the case of iron-containing catalysts, PG is adsorbed on the surface mainly in the bidentate configuration that contributes to the selective conversion of both hydroxyl groups with the formation of methylglyoxal. In the case of the Ag-containing catalyst, at low degree of PG conversion (up to 10%), only monodentate intermediates are adsorbed on the surface as evidenced by the high selectivity towards hydroxyacetone. As the contact time increases, PG is adsorbed on the surface of the Ag/SiO2 catalyst both in the mono- and bidentate configurations as evidenced by the methylglyoxal appearance in the products. However, an increase in the contact time also leads to the readsorption of products on the surface of the Ag/SiO2 catalyst, which is accompanied by high probability of the C–C bond cleavage; with an increase in the PG conversion, the selectivity towards C1 products sharply increases.
To substantiate the obtained experimental results, the quantum-chemical calculations of the main transformations of PG and oxygen into C
3 products were carried out accounting for the binding energies with the Fe-containing sites. The configuration of the active site for each catalyst was chosen based on the obtained phase analysis results and Raman spectroscopy results in Refs. [
13,
25]. FePO
4 and Fe
2(MoO
4)
3 structures were used as models of active sites in iron phosphate and iron molybdenum catalysts, respectively.
Results of theoretical calculations show that the first stage of reagent conversion comprises the adsorption and dissociation of molecular oxygen on the active sites of the catalysts (see
Supplementary Materials for the profile of interactions of the key reagents, intermediates (
Figure S4), and products with the active sites along the reaction coordinate (
Tables S1 and S2)). In both cases, the formation of atomic oxygen species adsorbed on Fe sites is observed. The PG is then adsorbed to form adsorbed PDO intermediate through the O–H bond breaking. Moreover, at the iron-phosphate site, a stronger interaction (−715 kJ/mol) occurs between the diol molecule and the Lewis Fe
3+ center contrary to the iron-molybdenum system (
Table 2). In this case, one should expect a decrease in the activity of the iron-phosphate catalyst due to an increase in the residence time of a strongly bound adsorption complex. In the case of the iron–molybdenum catalysts, the PG binding activates the O–H bond both on the Fe-containing sites and on the Fe-O-Mo moiety. The increase of the C–O bond length to up to 1.46 Å in the alcohol molecule (compared to 1.42 Å for the isolated molecule) also occurs at the Fe-containing sites.
The subsequent PDO conversion both at the FePO
4 and Fe
2(MoO
4)
3 sites occurs via the formation of intermediates, namely, propan-1-al (PPA) and oxopropoxy (OPO). In turn, the PPA and OPO are the intermediates species in the formation of methylglyoxal, hydroxyacetone (and formaldehyde), respectively. The binding energy of the PPA structure (
Table 2) is higher for the Fe
2(MoO
4)
3 site due to the assistance of adsorbed atomic oxygen species. The binding energies with the Fe
2(MoO
4)
3 site for the OPO and PPA intermediates are practically similar. However, the presence of the adsorbed oxygen atom strengthens the PPA binding energy with the catalyst surface, while for the OPO structure it results in its decrease. It is noteworthy that in the case of the Fe
2(MoO
4)
3 site, the strengthening of the binding energy with the catalyst surface occurs due to the breaking of the C
2–H bond followed by the methylglyoxal desorption and the formation of the Fe–O
adsH bond (O
ads—oxygen atom adsorbed on the Fe site).
As in the case of PDO intermediate formation, the strength of the OPO binding to the active site is higher for the FePO
4 site as compared to the Fe
2(MoO
4)
3 site (
Table 2). It is noteworthy that the OPO interaction with both FePO
4 and Fe
2(MoO
4)
3 sites is more favorable in the absence of adsorbed atomic oxygen species. Further transformation of the OPO at the Fe
2(MoO
4)
3 site in the presence of atomic oxygen is accompanied by the elongation of the C
1–C
2 bond to up to 1.67 Å (1.59 Å in the isolated species) followed by its cleavage and desorption of the C
1 products due to the weak interaction with the active site. In this case, the parallel transformations of the OPO intermediate into hydroxyacetone and C
1 by-products occur depending on the presence of the adsorbed oxygen atom. Moreover, at the FePO
4 sites, both transformation routes of the OPO intermediate are implemented to similar extent that is also evidenced by the experimental data (
Figure 2).
The methylglyoxal formation is more favorable during the PPA dehydrogenation, while hydroxyacetone is formed as a result of the OPO transformations through the oxidative route. Since the reduction of the surface sites in the H
2-TPR mode for the Fe
2(MoO
4)
3 catalyst [
25] proceeds at higher temperatures (by 100 °C higher as compared to FePO
4 [
24]) indicating higher strength of oxygen binding to the surface, the dehydrogenation route is more favorable. In the presence of the adsorbed oxygen atom, a competing process for the OPO selective oxidation to HA is the C–C bond cleavage in the OPO species yielding formaldehyde. At the same time, it is noteworthy that the oxygen presence in the reaction medium favorably affects the course of the oxidative processes as was shown in Ref. [
29] exemplified by the oxidation of organic compounds (acrolein, formaldehyde, ethanol, etc.) on the oxide V–Ti catalysts.
Thus, the studied systems are characterized by a relatively similar position of the main intermediates formed during the PG oxidation. However, significant differences are found in the interaction energies of the key PDO intermediate with the active sites of the FePO4 and Fe2(MoO4)3. The PDO binding energy on the iron-phosphate site is much lower and amounts to −586 kJ/mol (−715.5 kJ/mol in the presence of atomic oxygen). In this case, the strong PDO interaction with the FePO4 site leads to the increase of the residence time of the adsorbed intermediates on the active sites of the catalyst and, accordingly, to a decrease in the degree of PG conversion, which is consistent with the catalytic experiments. According to the results of theoretical calculations, on the iron-molybdenum site, an intermediate value of the PDO binding energy was obtained as compared to those over FePO4. Thus, it can be assumed that the use of iron-molybdenum catalysts leads to an increase in the turnover frequency for the Fe-containing sites upon interaction with the intermediate compounds and, at the same time, facilitates further selective oxidation of PG without breaking the C–C bond.

 ,
, 



{kind=link}
{kind=link}