
Surface and Structural Characterization of PVTMS Films Treated by Elemental Fluorine in Liquid Perfluorodecalin

 , ,
, ,  and
and
Abstract
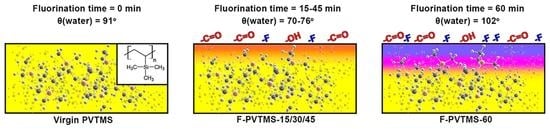
:
1. Introduction
2. Materials and Methods
2.1. Preparation of PVTMS Samples for Fluorination
2.2. Direct Liquid-Phase Fluorination of PVTMS Samples
2.3. X-ray Photoelectron Spectroscopy (XPS)
2.4. Scanning and Energy-Dispersive X-ray Spectroscopy (SEM and XEDS)
2.5. Attenuated Total Reflectance Infra-Red Spectroscopy (ATR-IR)
2.6. Contact Angle Measurements and Surface Energy Calculation
3. Results and Discussion
3.1. ATR-IR Analysis of the PVTMS Samples
3.2. XPS Analysis of the PVTMS Samples
3.3. SEM and XEDS for the PVTMS Samples
3.4. Contact Angle Measurements and Analysis of the Fluorinated PVTMS Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yampolskii, Y.P.; Durgarjan, S.G.; Nametkin, N.S. Permeability, Diffusion and Solubility of n-Alkanes in Polymers. Vysokomol. Soedin. B 1979, 21, 616–621. [Google Scholar]
- Robeson, L.M. The Upper Bound Revisited. J. Membr. Sci. 2008, 320, 390–400. [Google Scholar] [CrossRef]
- Bernardo, P.; Drioli, E.; Golemme, G. Membrane Gas Separation: A Review/State of the Art. Ind. Eng. Chem. Res. 2009, 48, 4638–4663. [Google Scholar] [CrossRef]
- Wang, Y.; Ghanem, B.S.; Han, Y.; Pinnau, I. State-of-the-Art Polymers of Intrinsic Microporosity for High-Performance Gas Separation Membranes. Curr. Opin. Chem. Eng. 2022, 35, 100755. [Google Scholar] [CrossRef]
- Shalygin, M.G.; Kozlova, A.A.; Netrusov, A.I.; Teplyakov, V.V. Vapor-Phase Membrane Concentration of Bioethanol and Biobutanol Using Hydrophobic Membranes Based on Glassy Polymers. Pet. Chem. 2016, 56, 977–986. [Google Scholar] [CrossRef]
- Koros, W.J.; Stannett, V.T.; Hopfenberg, H.B. Estimation of the Effective Permeability of Thin Surface Layers Created by Exposure of Polyethylene to Fluorine. Polym. Eng. Sci. 1982, 22, 22–738. [Google Scholar] [CrossRef]
- Liu, G.; Jin, W. Pervaporation Membrane Materials: Recent Trends and Perspectives. J. Membr. Sci. 2021, 636, 119557. [Google Scholar] [CrossRef]
- Adamson, A.W.; Gast, A.P. Physical Chemistry of Surfaces, 6th ed.; Wiley: New York, NY, USA, 1997; ISBN 978-0-471-14873-9. [Google Scholar]
- de Gennes, P.G. Wetting: Statics and Dynamics. Rev. Mod. Phys. 1985, 57, 827–863. [Google Scholar] [CrossRef]
- Starannikova, L.E.; Teplyakov, V.V.; Durgar’yan, S.G. Gas Permeability of Polyvinyltrimethylsilane Modified by the Radiation Grafting of Acrylonitrile. Polym. Sci. USSR 1986, 28, 1411–1416. [Google Scholar] [CrossRef]
- Osipov, O.A.; Slovetskii, D.I. Improvement in Gas Permeability, Selectivity, and Stability of Asymmetric Membranes Based on Poly(vinyl Trimethylsilane). Vysokomol. Soedin. B 1995, 37, 715–717. [Google Scholar]
- Syrtsova, D.A.; Shalygin, M.G.; Teplyakov, V.V.; Palanivelu, K.; Zinoviev, A.V.; Piskarev, M.S.; Kuznetsov, A.A. Enhancement of Gas Separation Properties of Polyvinyltrimethylsilane by Low-Temperature Plasma Treatment for Carbon Dioxide Utilization in “Green Chemistry” Processes. Membr. Membr. Technol. 2021, 3, 43–51. [Google Scholar] [CrossRef]
- Syrtsova, D.; Piskarev, M.; Zinoviev, A.; Kuznetsov, A.; Skryleva, E.; Gilman, A.; Teplyakov, V. The Gas Permeability Properties of Poly(Vinyltrimethylsilane) Treated by Low-temperature Plasma. J. Appl. Polym. Sci. 2022, 139, e52821. [Google Scholar] [CrossRef]
- Le Roux, J.D.; Teplyakov, V.V.; Paul, D.R. Gas Transport Properties of Surface Fluorinated Poly (Vinyltrimethylsilane) Films and Composite Membranes. J. Membr. Sci. 1994, 90, 55–68. [Google Scholar] [CrossRef]
- Kharitonov, A.P.; Moskvin, Y.L.; Teplyakov, V.V.; Le Roux, J.D. Direct Fluorination of Poly(Vinyl Trimethylsilane) and Poly(Phenylene Oxide). J. Fluor. Chem. 1999, 93, 129–137. [Google Scholar] [CrossRef]
- Syrtsova, D.A.; Kharitonov, A.P.; Teplyakov, V.V.; Koops, G.-H. Improving Gas Separation Properties of Polymeric Membranes Based on Glassy Polymers by Gas Phase Fluorination. Desalination 2004, 163, 273–279. [Google Scholar] [CrossRef]
- Belov, N.A.; Alentiev, A.Y.; Bogdanova, Y.G.; Vdovichenko, A.Y.; Pashkevich, D.S. Direct Fluorination as Method of Improvement of Operational Properties of Polymeric Materials. Polymers 2020, 12, 2836. [Google Scholar] [CrossRef]
- Cheng, Z.; Wu, P.; Li, B.; Chen, T.; Liu, Y.; Ren, M.; Wang, Z.; Lai, W.; Wang, X.; Liu, X. Surface Chain Cleavage Behavior of PBIA Fiber Induced by Direct Fluorination. Appl. Surf. Sci. 2016, 384, 480–486. [Google Scholar] [CrossRef]
- Cheng, Z.; Li, B.; Huang, J.; Chen, T.; Liu, Y.; Wang, X.; Liu, X. Covalent Modification of Aramid Fibers’ Surface via Direct Fluorination to Enhance Composite Interfacial Properties. Mater. Des. 2016, 106, 216–225. [Google Scholar] [CrossRef]
- Piskarev, M.; Skryleva, E.; Gilman, A.; Senatulin, B.; Zinoviev, A.; Syrtsova, D.; Teplyakov, V.; Kuznetsov, A. Depth Profile Analysis of the Modified Layer of Poly(Vinyltrimethylsilane) Films Treated by Direct-Current Discharge. Coatings 2021, 11, 1317. [Google Scholar] [CrossRef]
- Kloubek, J. Development of Methods for Surface Free Energy Determination Using Contact Angles of Liquids on Solids. Adv. Colloid Interface Sci. 1992, 38, 99–142. [Google Scholar] [CrossRef]
- Lee, L.-H. Correlation between Lewis Acid−Base Surface Interaction Components and Linear Solvation Energy Relationship Solvatochromic α and β Parameters. Langmuir 1996, 12, 1681–1687. [Google Scholar] [CrossRef]
- Blinov, I.A.; Mukhortov, D.A.; Yampolskii, Y.P.; Belov, N.A.; Alentiev, A.Y.; Chirkov, S.V.; Bondarenko, G.N.; Kostina, Y.V.; Legkov, S.A.; Perepuchov, A.M.; et al. Direct Fluorination of Poly-2,6-Dimethyl-1,4-Phenylene Oxide in Perfluorinated Liquid Medium. J. Fluor. Chem. 2020, 234, 109526. [Google Scholar] [CrossRef]
- Belov, N.A.; Nikiforov, R.Y.; Alentiev, A.Y.; Bezgin, D.A.; Blinov, I.A.; Suvorov, A.V.; Kostina, J.V.; Legkov, S.A.; Levin, I.S.; Gringolts, M.L.; et al. Gas Transport and Separation Properties of Polynorbornene Treated with Elemental Fluorine in a Perfluorodecalin Liquid. Membr. Membr. Technol. 2021, 3, 351–364. [Google Scholar] [CrossRef]
- Yancey, D.F.; Reinhardt, C. Damage and Repair of Organic and Inorganic Surfaces by Ar+ Ion and Gas Cluster Ion Beam Sputtering. J. Electron Spectrosc. Relat. Phenom. 2019, 231, 104–108. [Google Scholar] [CrossRef]
- Hofstetter, Y.J.; Vaynzof, Y. Quantifying the Damage Induced by X-ray Photoelectron Spectroscopy Depth Profiling of Organic Conjugated Polymers. ACS Appl. Polym. Mater. 2019, 1, 1372–1381. [Google Scholar] [CrossRef]
- Belov, N.A.; Blinov, I.A.; Suvorov, A.V.; Nikiforov, R.Y.; Chirkov, S.V.; Alentiev, A.Y.; Kambur, M.P.; Kostina, Y.V.; Levin, I.S.; Shapagin, A.V.; et al. Gas Permeability of Cellulose Acetate Films Treated with Fluorine in Perfluorodecalin. Membr. Membr. Technol. 2021, 3, 114–123. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equation | A | B, μm | C0, at. % |
|---|---|---|---|
| (1) | 0.006 μm/min | - | 1.41 |
| (2) | 0.064 at. %/min | 0.551 | 1.46 |
| Fluorination Time, min | θ(water), deg | θ(CH2I2), deg | γdSV, mJ/m2 | γpSV, mJ/m2 | γSV, mJ/m2 |
|---|---|---|---|---|---|
| 0 | 91 | 66 | 22 | 4 | 26 |
| 15 | 76 | 48 | 30 | 7 | 37 |
| 30 | 74 | 42 | 33 | 7 | 40 |
| 45 | 70 | 42 | 32 | 10 | 42 |
| 60 | 102 | 49 | 36 | 0 | 36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belov, N.A.; Alentiev, A.Y.; Pashkevich, D.S.; Voroshilov, F.A.; Dvilis, E.S.; Asanov, I.P.; Nikiforov, R.Y.; Chirkov, S.V.; Syrtsova, D.A.; Kostina, J.V.; et al. Surface and Structural Characterization of PVTMS Films Treated by Elemental Fluorine in Liquid Perfluorodecalin. Materials 2023, 16, 913. https://doi.org/10.3390/ma16030913
Belov NA, Alentiev AY, Pashkevich DS, Voroshilov FA, Dvilis ES, Asanov IP, Nikiforov RY, Chirkov SV, Syrtsova DA, Kostina JV, et al. Surface and Structural Characterization of PVTMS Films Treated by Elemental Fluorine in Liquid Perfluorodecalin. Materials. 2023; 16(3):913. https://doi.org/10.3390/ma16030913
Chicago/Turabian StyleBelov, Nikolay A., Aleksandr Y. Alentiev, Dmitrii S. Pashkevich, Fedor A. Voroshilov, Edgar S. Dvilis, Igor P. Asanov, Roman Y. Nikiforov, Sergey V. Chirkov, Daria A. Syrtsova, Julia V. Kostina, and et al. 2023. "Surface and Structural Characterization of PVTMS Films Treated by Elemental Fluorine in Liquid Perfluorodecalin" Materials 16, no. 3: 913. https://doi.org/10.3390/ma16030913
APA StyleBelov, N. A., Alentiev, A. Y., Pashkevich, D. S., Voroshilov, F. A., Dvilis, E. S., Asanov, I. P., Nikiforov, R. Y., Chirkov, S. V., Syrtsova, D. A., Kostina, J. V., & Bogdanova, Y. G. (2023). Surface and Structural Characterization of PVTMS Films Treated by Elemental Fluorine in Liquid Perfluorodecalin. Materials, 16(3), 913. https://doi.org/10.3390/ma16030913