
Development and Characterization of Innovative Multidrug Nanoformulation for Cardiac Therapy

 , , ,
, , , 
Abstract
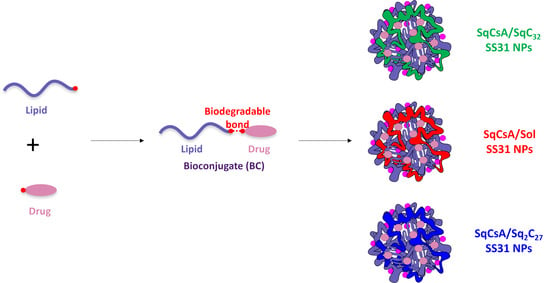
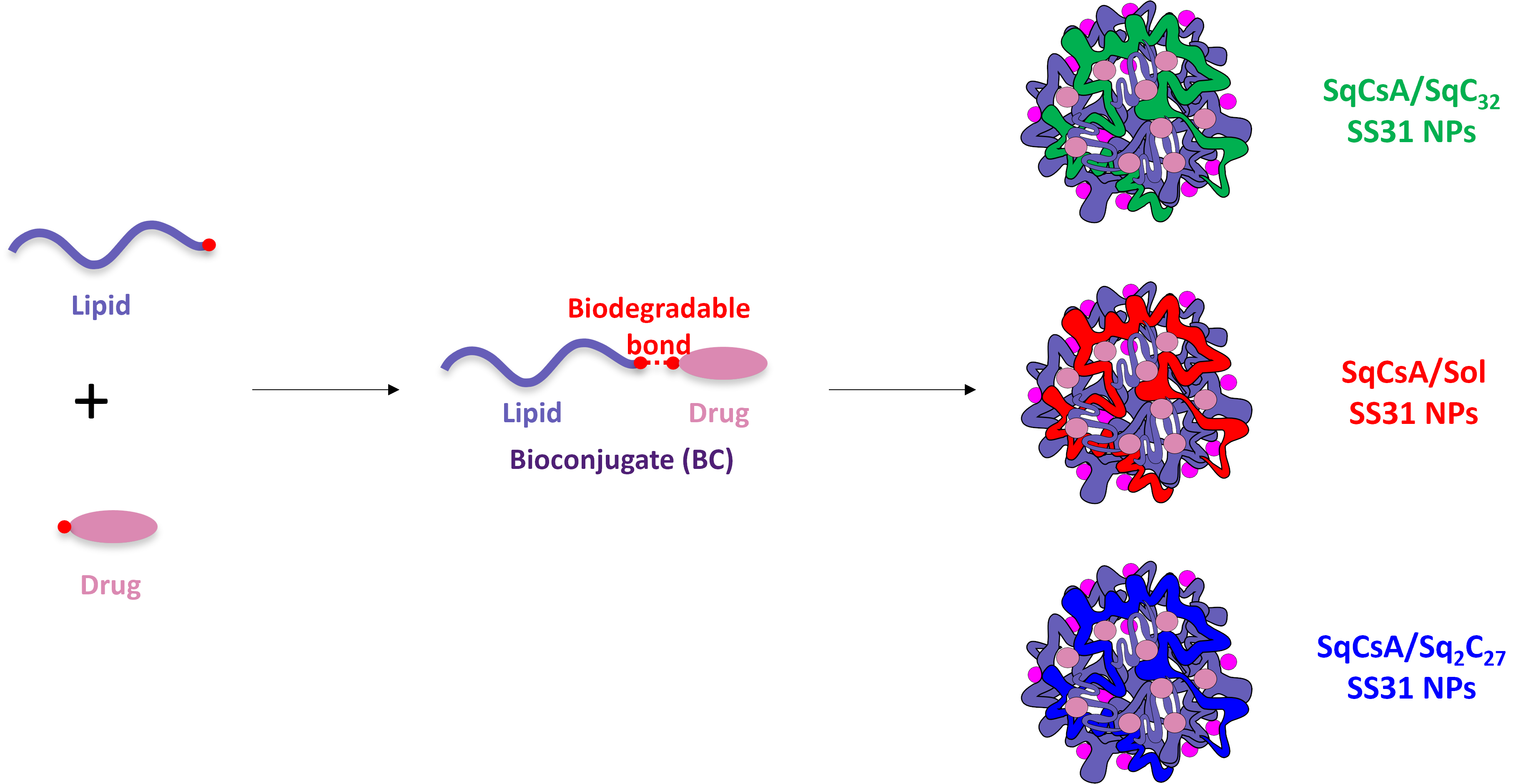
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. NMR Data of the Peptides
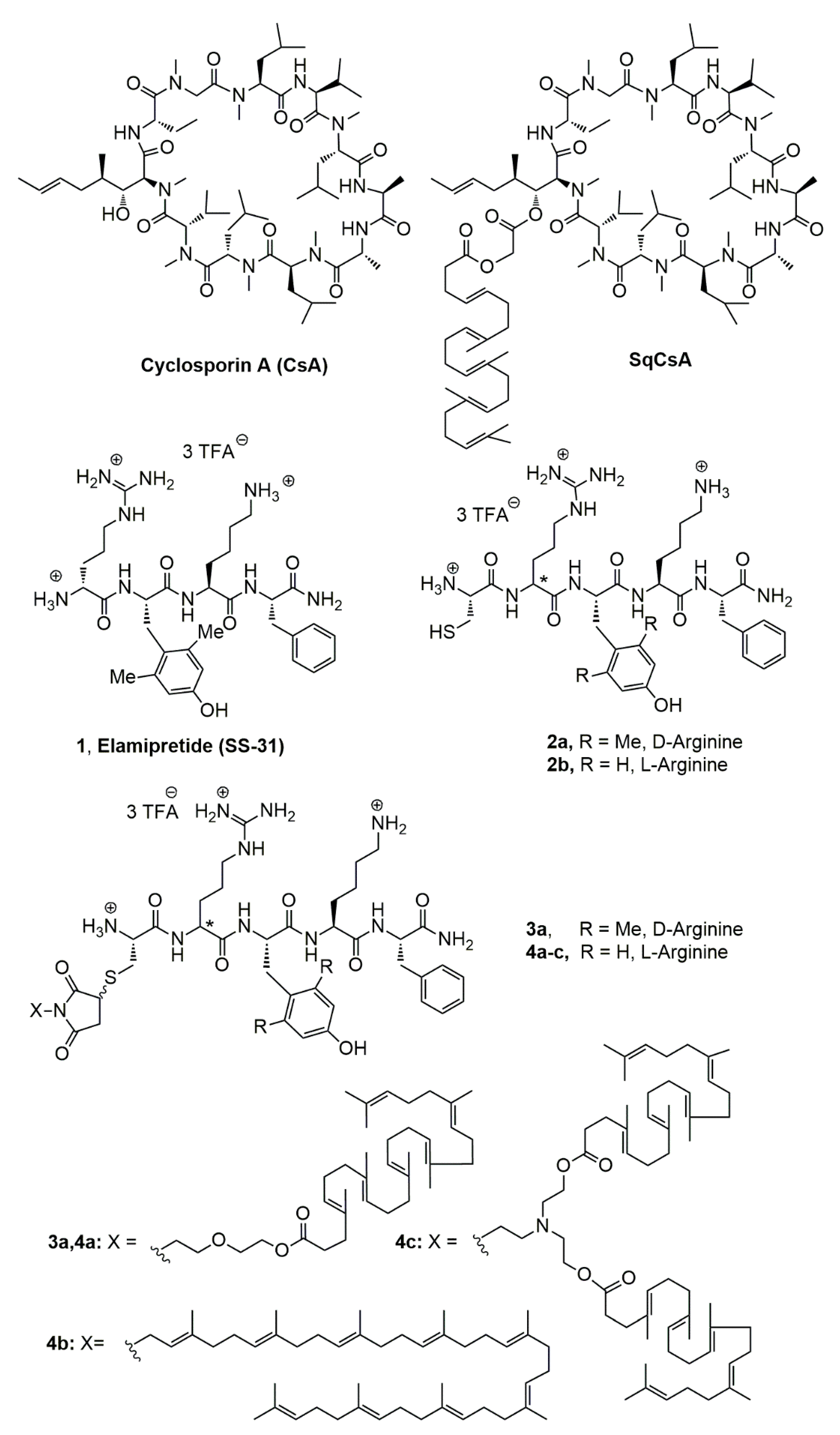
2.2.1. NMR Data of [Cys-Arg-Tyr-Lys-Phe-NH2]3+ 3CF3CO2− (2b, Figure 1)
2.2.2. NMR Data of [Cys-D-Arg-diMeTyr-Lys-Phe-NH2]3+ 3CF3CO2− (2a, Figure 1)
2.3. Synthesis of the Lipid Peptide Bioconjugates
2.3.1. General
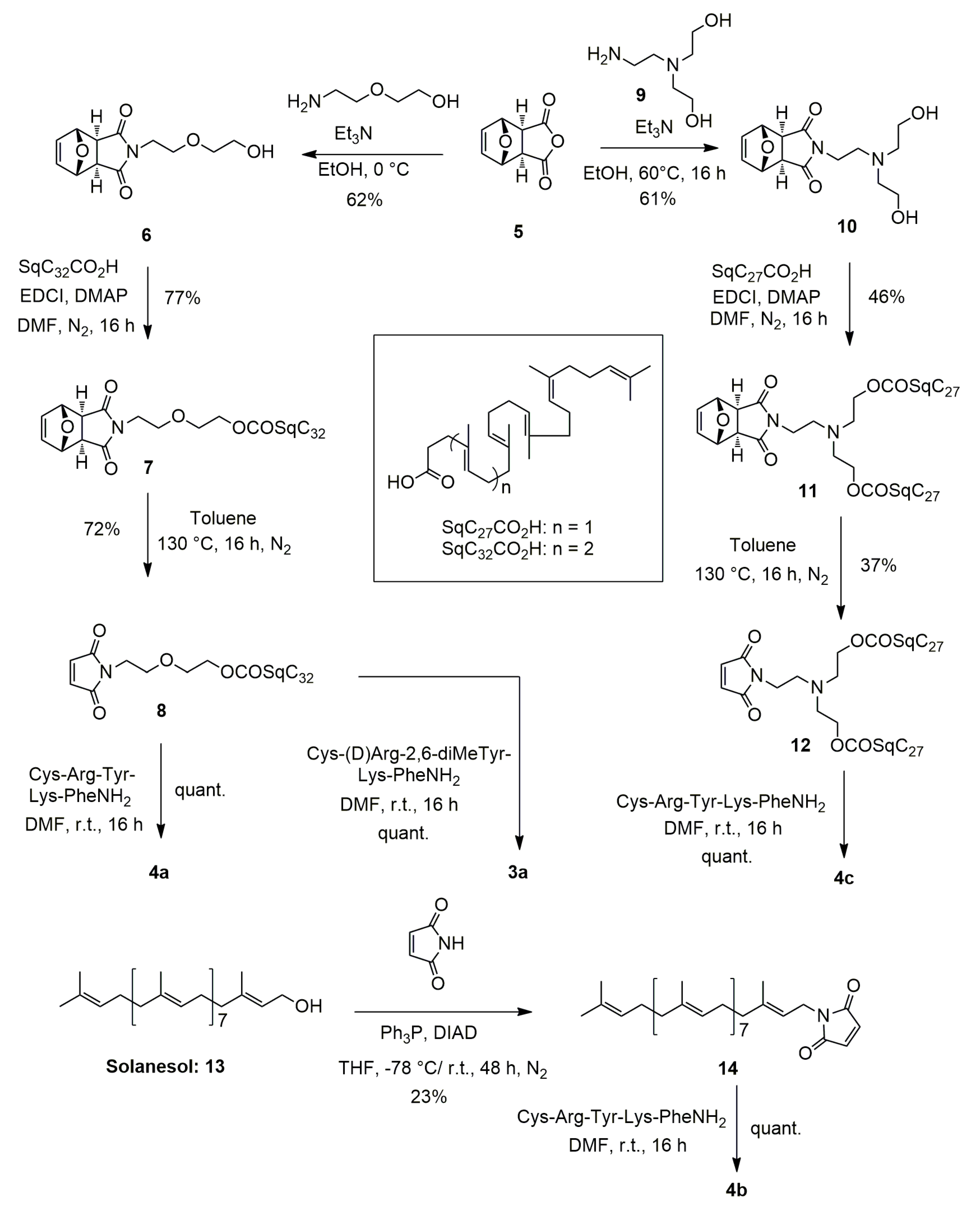
2.3.2. Synthesis of 4-[2-(2-Hydroxyethoxy)ethyl]-10-oxa-4-azatricyclo [5.2.1.02,6]dec-8-ene-3,5-dione (6) (Figure 2)

2.3.3. Synthesis of 2-(2-{3,5-dioxo-10-oxa-4-azatricyclo [5.2.1.02,6]dec-8-en-4-yl}ethoxy)ethyl (4E,8E,12E,16E,20E)-4,8,12,17,21,25-hexamethylhexacosa-4,8,12,16,20,24-hexaenoate (7) (Figure 2)
2.3.4. Synthesis of 2-[2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethoxy]ethyl (4E,8E,12E,16E,20E)-4,8,12,17,21,25-hexamethylhexacosa-4,8,12,16,20,24-hexaenoate (8)
2.3.5. Synthesis of Squalene Acetic Acid Conjugate of Cys-Arg-Tyr-Lys-PheNH2 (4a)
2.3.6. Synthesis of 1-[(2E,6E,10E,14E,18E,22E,26E,30E)-3,7,11,15,19,23,27,31,35-nonamethylhexatria-conta-2,6,10,14,18,22,26,30,34-nonaen-1-yl]-2,5-dihydro-1H-pyrrole-2,5-dione (14)
2.3.7. Synthesis of Solanesol Conjugate of Cys-Arg-Tyr-Lys-Phe-NH2 (4b)
2.3.8. Synthesis of 4-{2-[bis(2-hydroxyethyl)amino]ethyl}-10-oxa-4-azatricyclo [5.2.1.02,6]dec-8-ene-3,5-dione (10)
2.3.9. Synthesis of 2-{[2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl](2-{[(4E,8E,12E,16E)-4,8,12,17,21-pentamethyldocosa-4,8,12,16,20-pentaenoyl]oxy}ethyl) amino}ethyl (4E,8E,12E,16E)-4,8,12,17,21-pentamethyldocosa-4,8,12,16,20-pentaenoate (12)
2.3.10. Synthesis of Bis-Squalene Conjugate of Cys-Arg-Tyr-Lys-PheNH2 (4c)
2.3.11. Synthesis of Squalene Acetic Acid Conjugate of Cys-D-Arg-diMeTyr-Lys-Phe-NH2 (3a)
2.4. Nanoparticle Obtention and Characterisation
2.5. Morphology by CryoTEM
2.6. X-ray Photoelectron Spectroscopy (XPS)
2.7. Cell Culture
2.8. Cytotoxicity of SqCsA/4a, 4b or 4c NPs
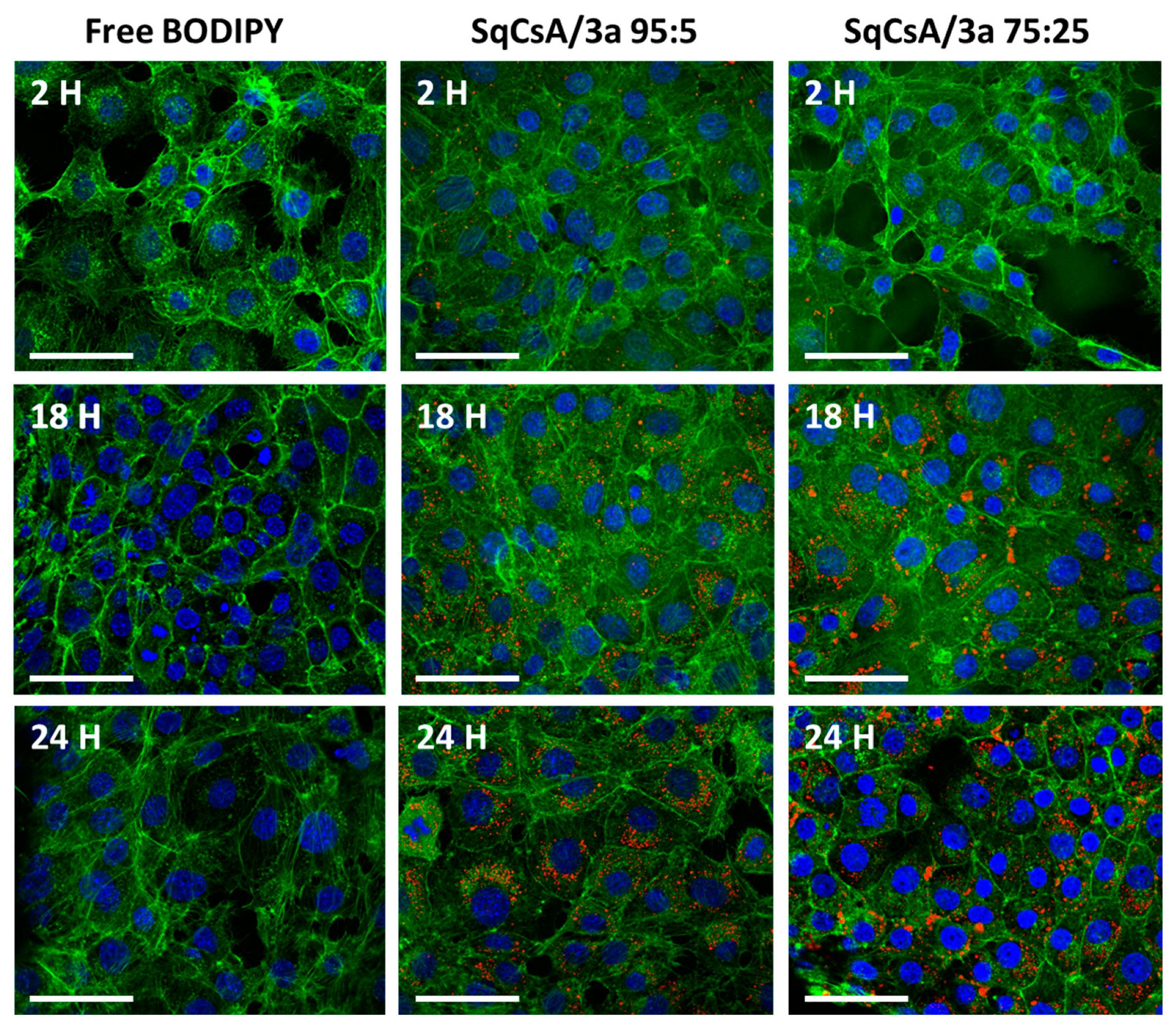
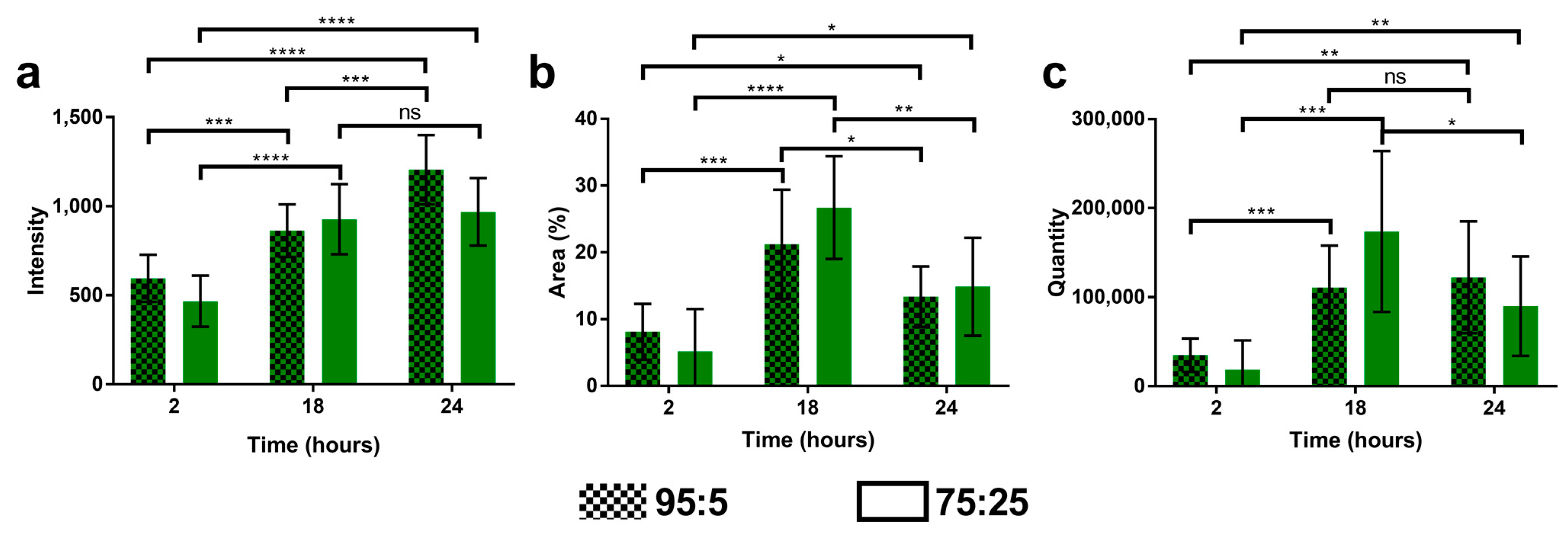
2.9. Cell Uptake of Multidrug Nanoparticles
2.10. Determination of Antioxidant Capacity
2.11. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of the Lipid-Cys-RYKF Bioconjugates
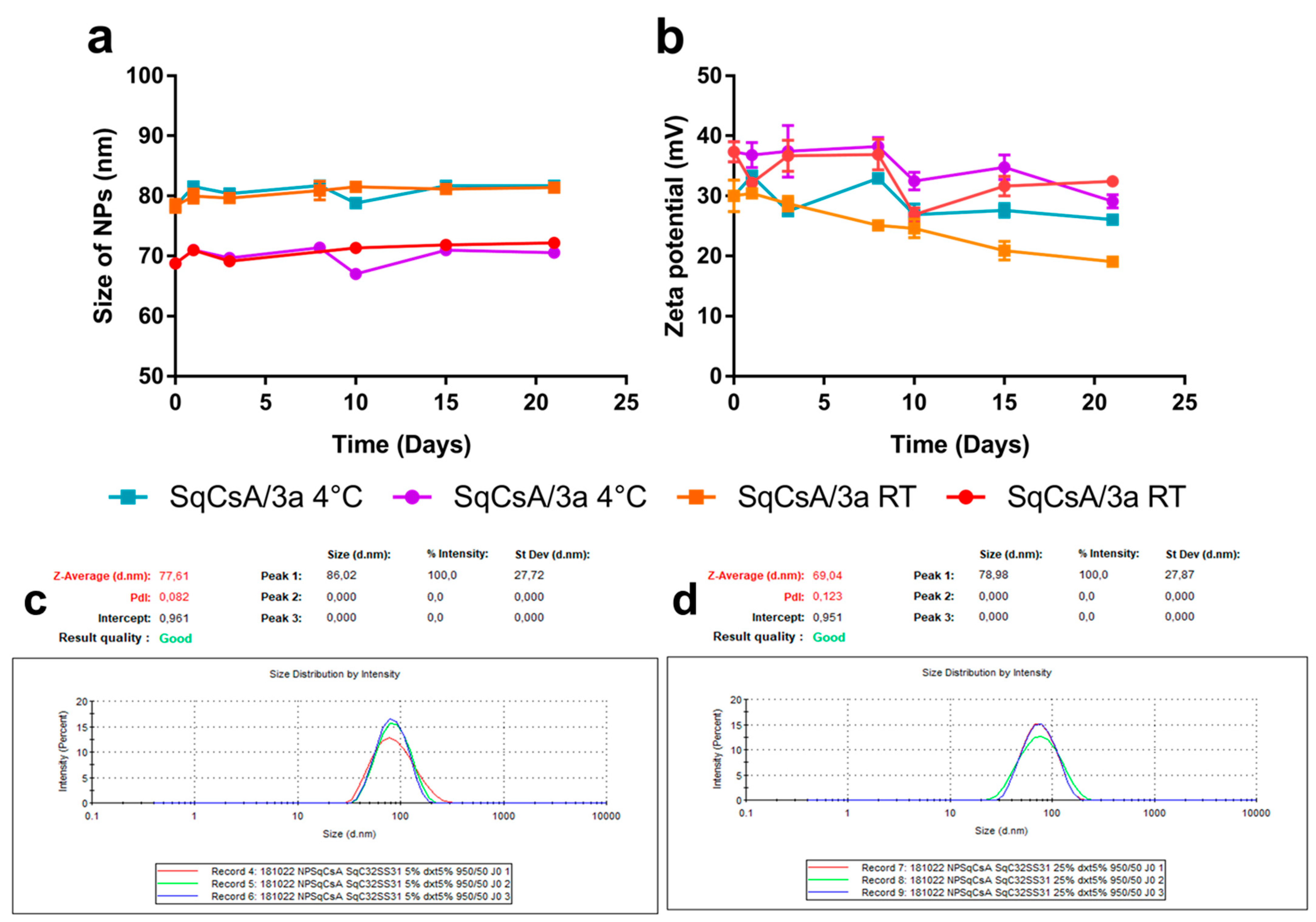
3.2. Nanoparticle Obtention
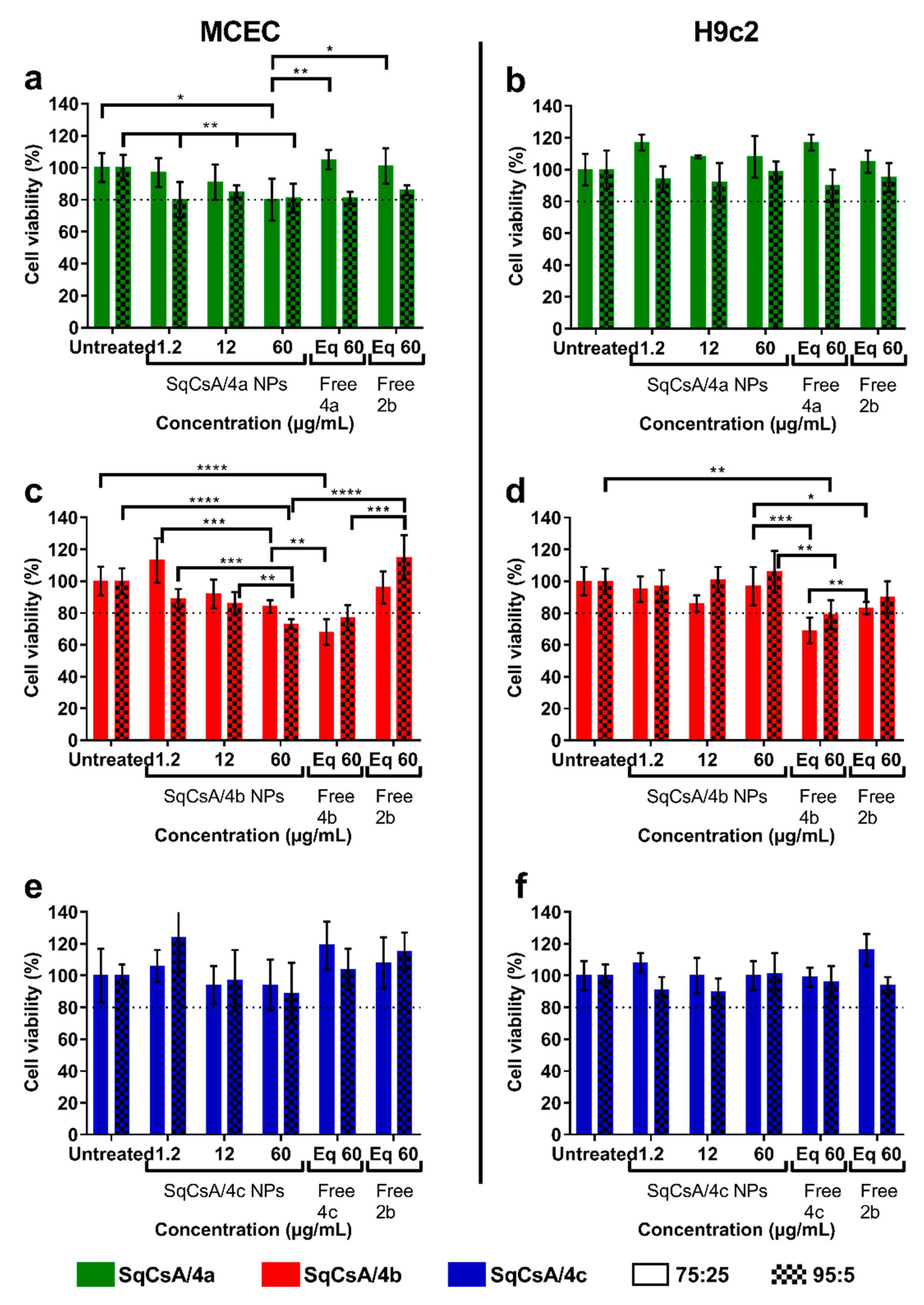
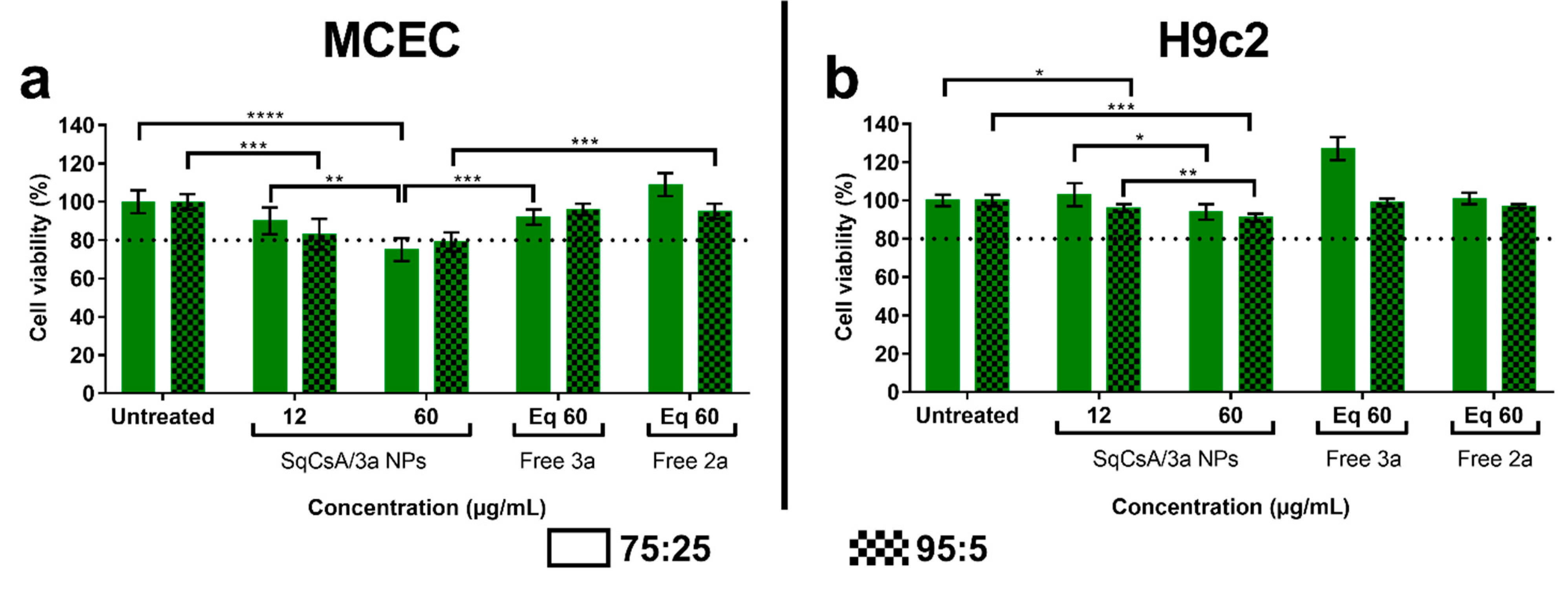
3.3. Cytotoxicity of SqCsA/4a, 4b and 4c NPs
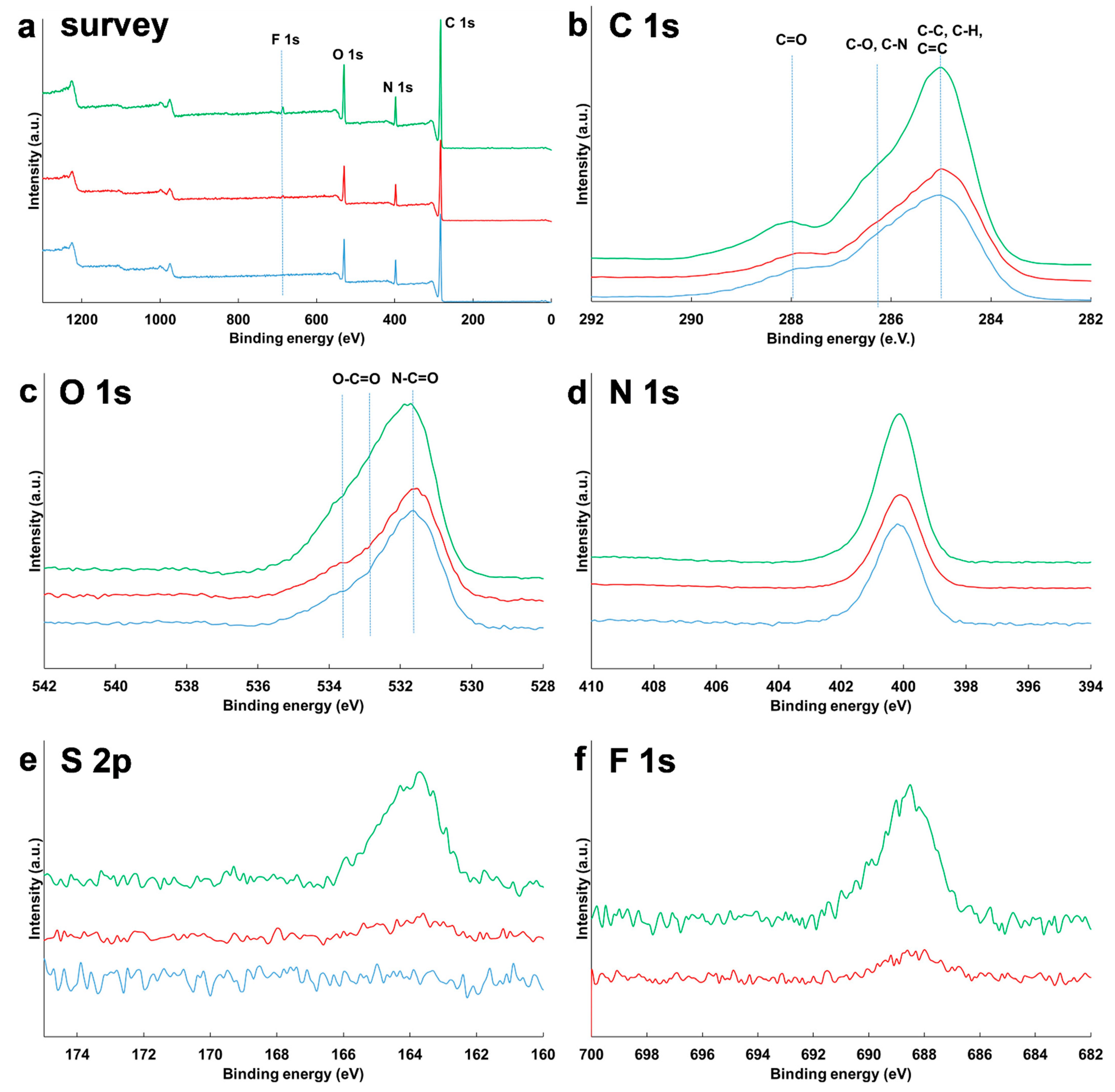
3.4. Surface Composition of NPs
3.5. Cell Uptake of Multidrug Nanoparticles
3.6. Antioxidant Capacity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hausenloy, D.J.; Yellon, D.M. Myocardial Ischemia-Reperfusion Injury: A Neglected Therapeutic Target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Fernandez Rico, C.; Konate, K.; Josse, E.; Nargeot, J.; Barrère-Lemaire, S.; Boisguérin, P. Therapeutic Peptides to Treat Myocardial Ischemia-Reperfusion Injury. Front. Cardiovasc. Med. 2022, 9, 792885. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.-M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-Permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane Inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. Cell-Permeable, Mitochondrial-Targeted, Peptide Antioxidants. AAPS J. 2006, 8, E277–E283. [Google Scholar] [CrossRef]
- Cho, J.; Won, K.; Wu, D.; Soong, Y.; Liu, S.; Szeto, H.H.; Hong, M.K. Potent Mitochondria-Targeted Peptides Reduce Myocardial Infarction in Rats. Coron. Artery Dis. 2007, 18, 215–220. [Google Scholar] [CrossRef]
- Zhao, K.; Luo, G.; Giannelli, S.; Szeto, H.H. Mitochondria-Targeted Peptide Prevents Mitochondrial Depolarization and Apoptosis Induced by Tert-Butyl Hydroperoxide in Neuronal Cell Lines. Biochem. Pharmacol. 2005, 70, 1796–1806. [Google Scholar] [CrossRef]
- Kloner, R.A.; Hale, S.L.; Dai, W.; Gorman, R.C.; Shuto, T.; Koomalsingh, K.J.; Gorman, J.H.; Sloan, R.C.; Frasier, C.R.; Watson, C.A.; et al. Reduction of Ischemia/Reperfusion Injury With Bendavia, a Mitochondria-Targeting Cytoprotective Peptide. J. Am. Heart Assoc. 2012, 1, e001644. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The Cardiolipin-Binding Peptide Elamipretide Mitigates Fragmentation of Cristae Networks Following Cardiac Ischemia Reperfusion in Rats. Commun. Biol. 2020, 3, 389. [Google Scholar] [CrossRef]
- Gibson, C.M.; Giugliano, R.P.; Kloner, R.A.; Bode, C.; Tendera, M.; Jánosi, A.; Merkely, B.; Godlewski, J.; Halaby, R.; Korjian, S.; et al. EMBRACE STEMI Study: A Phase 2a Trial to Evaluate the Safety, Tolerability, and Efficacy of Intravenous MTP-131 on Reperfusion Injury in Patients Undergoing Primary Percutaneous Coronary Intervention. Eur. Heart J. 2016, 37, 1296–1303. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. Mitochondria-Targeted Peptide Antioxidants: Novel Neuroprotective Agents. AAPS J. 2006, 8, E521–E531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-X.; Cheng, Y.; Liu, D.-Z.; Liu, M.; Cui, H.; Zhang, B.-L.; Mei, Q.-B.; Zhou, S.-Y. Mitochondria-Targeted Cyclosporin A Delivery System to Treat Myocardial Ischemia Reperfusion Injury of Rats. J. Nanobiotechnol. 2019, 17, 18. [Google Scholar] [CrossRef] [Green Version]
- Kuang, X.; Zhou, S.; Guo, W.; Wang, Z.; Sun, Y.; Liu, H. SS-31 Peptide Enables Mitochondrial Targeting Drug Delivery: A Promising Therapeutic Alteration to Prevent Hair Cell Damage from Aminoglycosides. Drug Deliv. 2017, 24, 1750–1761. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Jin, F.; Shu, G.; Xu, X.; Qi, J.; Kang, X.; Yu, H.; Lu, K.; Jiang, S.; Han, F.; et al. Enhanced Efficiency of Mitochondria-Targeted Peptide SS-31 for Acute Kidney Injury by PH-Responsive and AKI-Kidney Targeted Nanopolyplexes. Biomaterials 2019, 211, 57–67. [Google Scholar] [CrossRef]
- Qian, K.; Bao, X.; Li, Y.; Wang, P.; Guo, Q.; Yang, P.; Xu, S.; Yu, F.; Meng, R.; Cheng, Y.; et al. Cholinergic Neuron Targeting Nanosystem Delivering Hybrid Peptide for Combinatorial Mitochondrial Therapy in Alzheimer’s Disease. ACS Nano 2022, 16, 11455–11472. [Google Scholar] [CrossRef]
- Rai, S.; Singh, N.; Bhattacharya, S. Concepts on Smart Nano-Based Drug Delivery System. Recent Pat. Nanotechnol. 2021, 16, 67–89. [Google Scholar] [CrossRef]
- Yang, J.; Jia, C.; Yang, J. Designing Nanoparticle-Based Drug Delivery Systems for Precision Medicine. Int. J. Med. Sci. 2021, 18, 2943–2949. [Google Scholar] [CrossRef]
- Henderson, L.; Neumann, O.; Kadria-Vili, Y.; Gerislioglu, B.; Bankson, J.; Nordlander, P.; Halas, N.J. Plasmonic Gadolinium Oxide Nanomatryoshkas: Bifunctional Magnetic Resonance Imaging Enhancers for Photothermal Cancer Therapy. PNAS Nexus 2022, 1, pgac140. [Google Scholar] [CrossRef]
- Zhang, F.; Zhuang, J.; Li, Z.; Gong, H.; de Ávila, B.E.-F.; Duan, Y.; Zhang, Q.; Zhou, J.; Yin, L.; Karshalev, E.; et al. Nanoparticle-Modified Microrobots for in Vivo Antibiotic Delivery to Treat Acute Bacterial Pneumonia. Nat. Mater. 2022, 21, 1324–1332. [Google Scholar] [CrossRef]
- Gendron, A.; Lan Linh Tran, N.; Laloy, J.; Brusini, R.; Rachet, A.; Gobeaux, F.; Nicolas, V.; Chaminade, P.; Abreu, S.; Desmaële, D.; et al. New Nanoparticle Formulation for Cyclosporin A: In Vitro Assessment. Pharmaceutics 2021, 13, 91. [Google Scholar] [CrossRef]
- Othman, M.; Desmaële, D.; Couvreur, P.; Vander Elst, L.; Laurent, S.; Muller, R.N.; Bourgaux, C.; Morvan, E.; Pouget, T.; Lepêtre-Mouelhi, S.; et al. Synthesis and Physicochemical Characterization of New Squalenoyl Amphiphilic Gadolinium Complexes as Nanoparticle Contrast Agents. Org. Biomol. Chem. 2011, 9, 4367. [Google Scholar] [CrossRef]
- Woodward, R.B.; Baer, H. The Reaction of Furan with Maleic Anhydride. J. Am. Chem. Soc. 1948, 70, 1161–1162. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raouane, M.; Desmaele, D.; Gilbert-Sirieix, M.; Gueutin, C.; Zouhiri, F.; Bourgaux, C.; Lepeltier, E.; Gref, R.; Ben Salah, R.; Clayman, G.; et al. Synthesis, Characterization, and in Vivo Delivery of SiRNA-Squalene Nanoparticles Targeting Fusion Oncogene in Papillary Thyroid Carcinoma. J. Med. Chem. 2011, 54, 4067–4076. [Google Scholar] [CrossRef]
- Massaad-Massade, L.; Boutary, S.; Caillaud, M.; Gracia, C.; Parola, B.; Gnaouiya, S.B.; Stella, B.; Arpicco, S.; Buchy, E.; Desmaële, D.; et al. New Formulation for the Delivery of Oligonucleotides Using “Clickable” SiRNA-Polyisoprenoid-Conjugated Nanoparticles: Application to Cancers Harboring Fusion Oncogenes. Bioconjug. Chem. 2018, 29, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.A. A High Yielding Synthesis of N-Alkyl Maleimides Using a Novel Modification of the Mitsunobu Reaction. J. Org. Chem. 1995, 60, 5352–5355. [Google Scholar] [CrossRef]
- Pilet, O.; Vogel, P. Synthesis and Diels-Alder Reactivity of 2,3,5,6-Tetramethylidenenorbornane. Helv. Chim. Acta 1981, 64, 2563–2570. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Cosco, D.; Rocco, F.; Ceruti, M.; Vono, M.; Fresta, M.; Paolino, D. Self-Assembled Squalenoyl-Cytarabine Nanostructures as a Potent Nanomedicine for Treatment of Leukemic Diseases. Int. J. Nanomedicine 2012, 7, 2535–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valetti, S.; Mura, S.; Noiray, M.; Arpicco, S.; Dosio, F.; Vergnaud, J.; Desmaële, D.; Stella, B.; Couvreur, P. Peptide Conjugation: Before or After Nanoparticle Formation? Bioconjug. Chem. 2014, 25, 1971–1983. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lepetre-Mouelhi, S.; Gautier, A.; Mura, S.; Cailleau, C.; Coudore, F.; Hamon, M.; Couvreur, P. A New Painkiller Nanomedicine to Bypass the Blood-Brain Barrier and the Use of Morphine. Sci. Adv. 2019, 5, eaau5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Feng, M.; Wang, X.; Zhang, H.; Ding, J.; Cheng, Z.; Qian, L. Peptide Szeto-Schiller 31 Ameliorates Doxorubicin-induced Cardiotoxicity by Inhibiting the Activation of the P38 MAPK Signaling Pathway. Int. J. Mol. Med. 2021, 47, 63. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.; Tamucci, J.D.; Ng, E.L.; Liu, S.; Birk, A.V.; Szeto, H.H.; May, E.R.; Alexandrescu, A.T.; Alder, N.N. Structure- activity Relationships of Mitochondria- targeted Tetrapeptide Pharmacological Compounds. eLife 2022, 11, e75531. [Google Scholar] [CrossRef]
- Santonocito, D.; Sarpietro, M.G.; Carbone, C.; Panico, A.; Campisi, A.; Siciliano, E.A.; Sposito, G.; Castelli, F.; Puglia, C. Curcumin Containing PEGylated Solid Lipid Nanoparticles for Systemic Administration: A Preliminary Study. Molecules 2020, 25, 2991. [Google Scholar] [CrossRef]
- Salehi, B.; Del Prado-Audelo, M.L.; Cortés, H.; Leyva-Gómez, G.; Stojanović-Radić, Z.; Singh, Y.D.; Patra, J.K.; Das, G.; Martins, N.; Martorell, M.; et al. Therapeutic Applications of Curcumin Nanomedicine Formulations in Cardiovascular Diseases. JCM 2020, 9, 746. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ratios | Size (nm) | PDI | Zeta (mV) | Drug Loading (%) | |
|---|---|---|---|---|---|
| SqCsA/4a | 95:5 | 86 ± 31 | 0.129 | +43 ± 15 | 3.1 |
| 90:10 | 88 ± 31 | 0.124 | +37 ± 12 | 6.2 | |
| 75:25 | 79 ± 30 | 0.141 | +47 ± 16 | 15.6 | |
| SqCsA/4b | 95:5 | 68 ± 27 | 0.161 | +36 ± 11 | 3.0 |
| 90:10 | 68 ± 27 | 0.160 | +44 ± 12 | 6.0 | |
| 75:25 | 62 ± 27 | 0.183 | +36 ± 13 | 14.7 | |
| SqCsA/4c | 95:5 | 66 ± 29 | 0.195 | +35 ± 14 | 2.6 |
| 90:10 | 64 ± 29 | 0.208 | +54 ± 8 | 5.2 | |
| 75:25 | 56 ± 26 | 0.217 | +34 ± 38 | 12.9 | |
| SqCsA/3a | 95:5 | 78 ± 26 | 0.111 | +30 ± 8 | 3.2 |
| 75:25 | 69 ± 25 | 0.134 | +37 ± 5 | 15.8 |
| Type of Molecule | Ratio | ||
|---|---|---|---|
| 95:5 | 75:25 | ||
| SqCsA/3a NPs | 12 µg/mL | 0.107 | 10.392 |
| 60 µg/mL | 2.144 | 49.796 | |
| Free 3a (bioconjugate) | 10.715 | 47.557 | |
| Free 2a (peptide) | 14.065 | 66.667 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gendron, A.; Domenichini, S.; Zanna, S.; Gobeaux, F.; Piesse, C.; Desmaële, D.; Varna, M. Development and Characterization of Innovative Multidrug Nanoformulation for Cardiac Therapy. Materials 2023, 16, 1812. https://doi.org/10.3390/ma16051812
Gendron A, Domenichini S, Zanna S, Gobeaux F, Piesse C, Desmaële D, Varna M. Development and Characterization of Innovative Multidrug Nanoformulation for Cardiac Therapy. Materials. 2023; 16(5):1812. https://doi.org/10.3390/ma16051812
Chicago/Turabian StyleGendron, Amandine, Séverine Domenichini, Sandrine Zanna, Frédéric Gobeaux, Christophe Piesse, Didier Desmaële, and Mariana Varna. 2023. "Development and Characterization of Innovative Multidrug Nanoformulation for Cardiac Therapy" Materials 16, no. 5: 1812. https://doi.org/10.3390/ma16051812
APA StyleGendron, A., Domenichini, S., Zanna, S., Gobeaux, F., Piesse, C., Desmaële, D., & Varna, M. (2023). Development and Characterization of Innovative Multidrug Nanoformulation for Cardiac Therapy. Materials, 16(5), 1812. https://doi.org/10.3390/ma16051812