
3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Simulation Model and Method
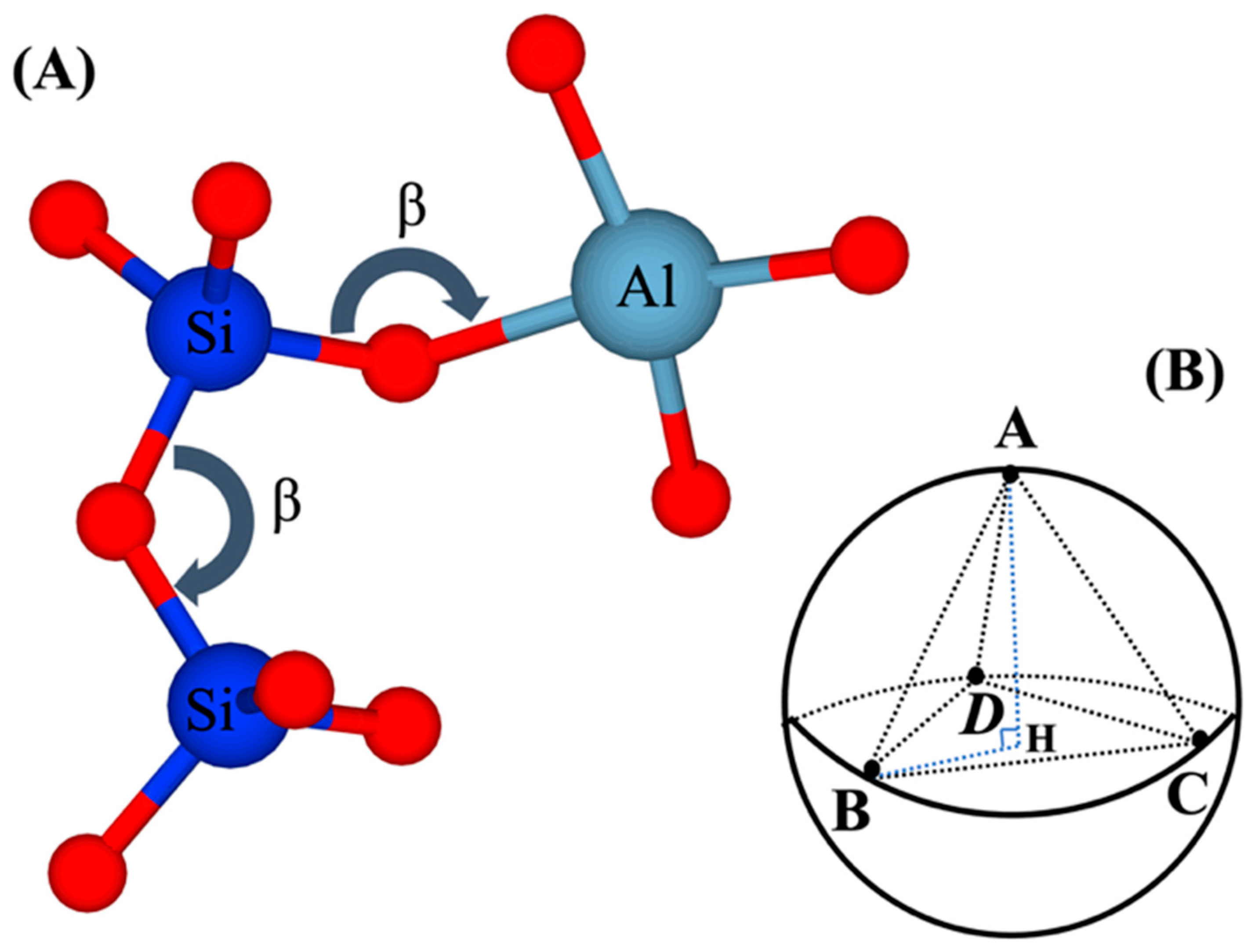
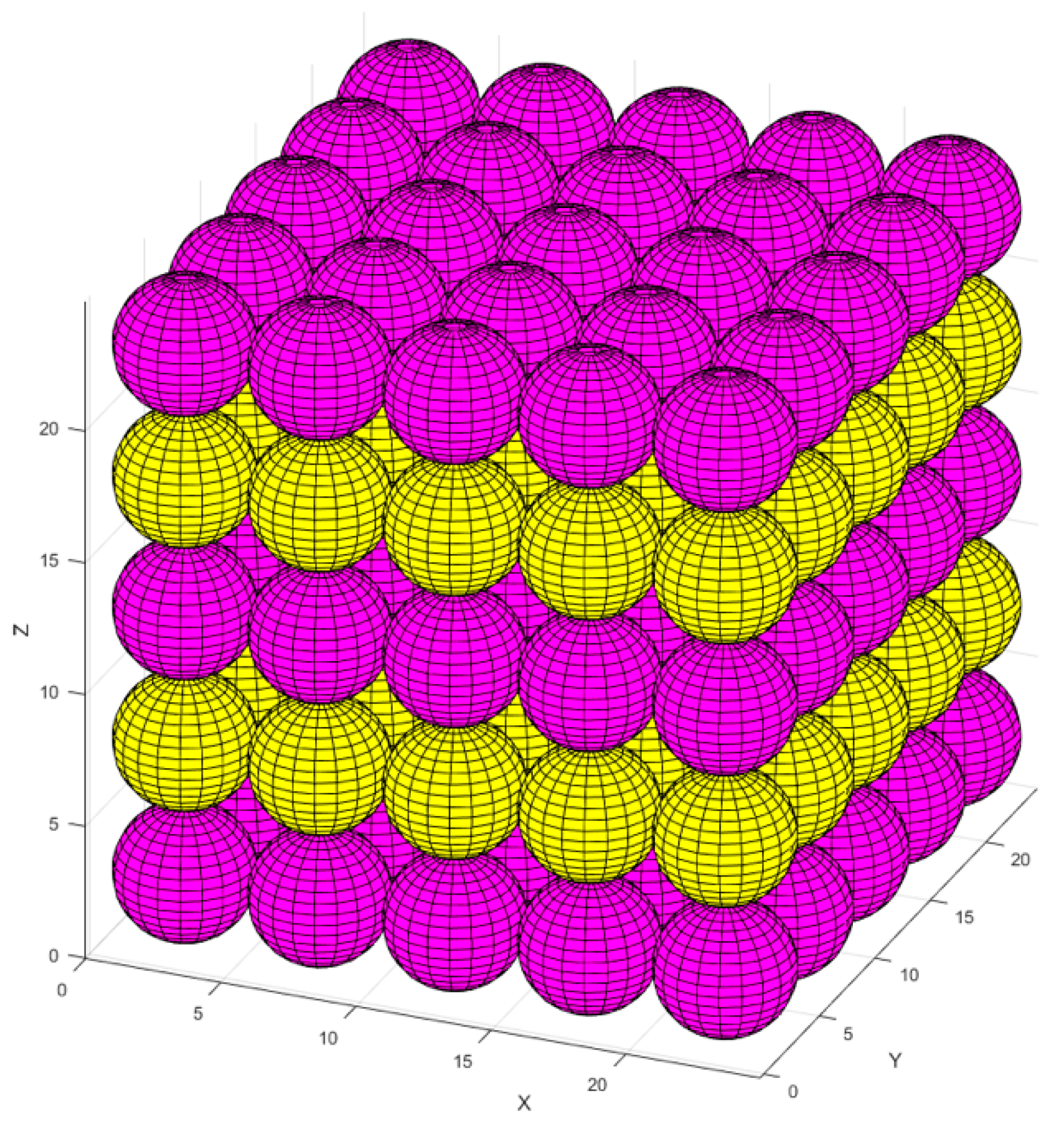
2.1. Atomistic Model Preparation
2.2. Monte Carlo Approach: Implementation in MATLAB Code
- (1)
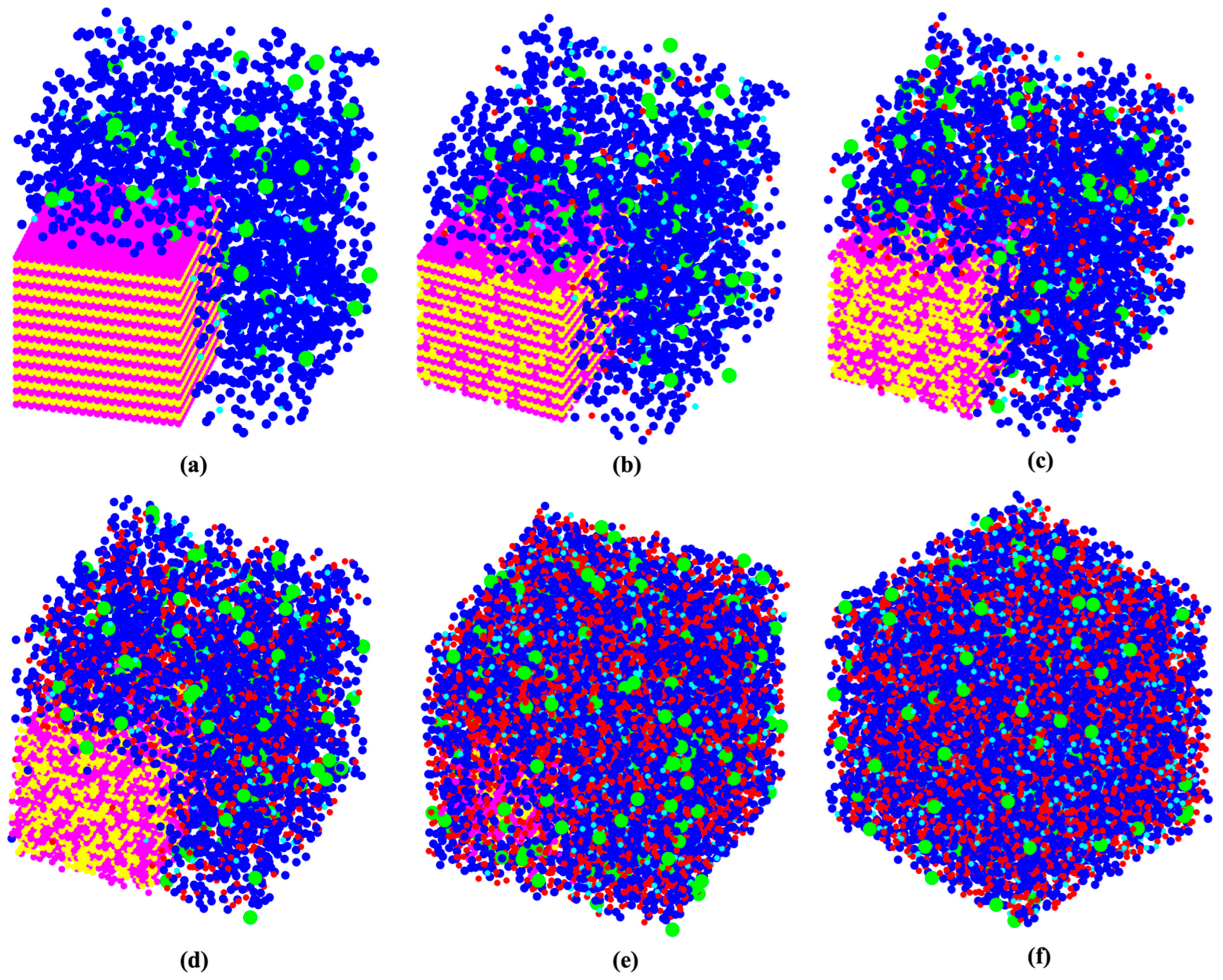
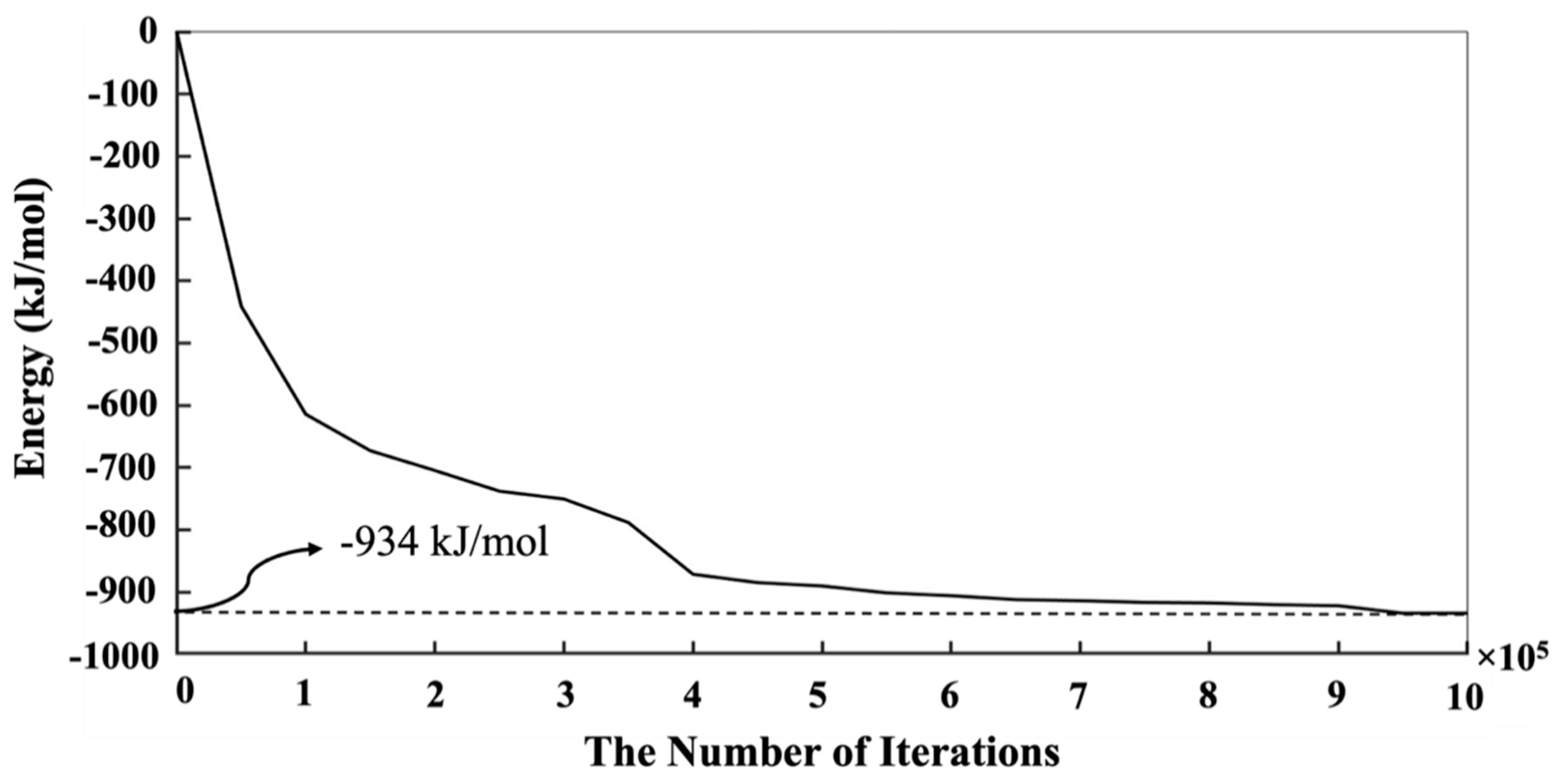
- First, we needed to simulate a silicate solution system that contained three types of dissolved silicate monomers (as particle types): Si(OH)4, SiO(OH)3−·Na+·3H2O, and SiO2(OH)22−·2Na+·6H2O. Particles were subjected to pre-equilibration of the energy of the system for 1 million iterations, where only MC moves involving particles not contained in the metakaolin system were allowed to be accepted. The particle movement through the MC approach was accepted if the system’s total energy was lower than the former system before the movement. On the contrary, if the system’s total energy was higher (less negative) than the former system before the particle movement, a movement for each iteration may have been accepted if the probability of X was higher than a selected random number between 0 and 1. The probability of X was computed based on the Boltzmann factor associated with the configurational change as described by Equation (1), where kB is the Boltzmann constant, T is the temperature, and ΔE is the change in energy.The dissolved three types of silicate particles in the solution were subjected to reaching the equilibrium condition (for our particular case, it took 1,000,000 iterations). Each iteration represented one random selection of a particle, including its movement to any arbitrary direction for a displacement, ensuring the particle connections always satisfied the tetrahedral structure. The displacement was generated by taking a random number from zero to one and multiplying it by the radius of a moving particle, and consequently determining whether the overlapping happened with the other particles. If yes, the particle went back to its previous position, and the next simulation loop was initiated. Finally, particles connected (into a cluster) when the distance between the moved particle and its nearest neighbor was within 1 Å; then, we calculated the increment/decrement of the system’s total energy [37]. Moreover, a tetrahedral geometry of the binding was respected, as described later (after Point 5 below).
- (2)
- Once the system’s total energy did not change, the solution reached equilibrium. The metakaolin sub-system was involved in the MC particle selection and movement process. Thus, the metakaolin sub-system’s random dissolution occurred from the outer surfaces exposed to the solution. After picking one surface particle from the metakaolin sub-system and dissolving it into the solution, the rejection and acceptance were checked. The rejection was regarded as the case of overlapping of dissolved metakaolin particles with other particles in the solution and an update of the total energy of the system based on the probability of X as mentioned in Equation (1). It is also worth mentioning that in the case of silicate being selected from the surface of the metakaolin sub-system, the silicate species type was also required to be specified, whose selection was based on the maintenance of the equilibration requirement. Then, another iteration process was carried out with the dissolved particles in the solution to perform MC movement for polymerization (i.e., particle binding/clustering).
- (3)
- It was essential to update the inner sites of the metakaolin system to become a part of the outer sites after each dissolution (particle removal) from the metakaolin system (after each dissolution iteration) and then continue the dissolution of metakaolin. Each dissolution process was followed by 30 MC iterations (i.e., particle movements) in the solution (Step 2).
- (4)
- Steps 2, 3, and 4 were carried out until the end of metakaolin dissolution, or until it could not be dissolved anymore.
- (5)
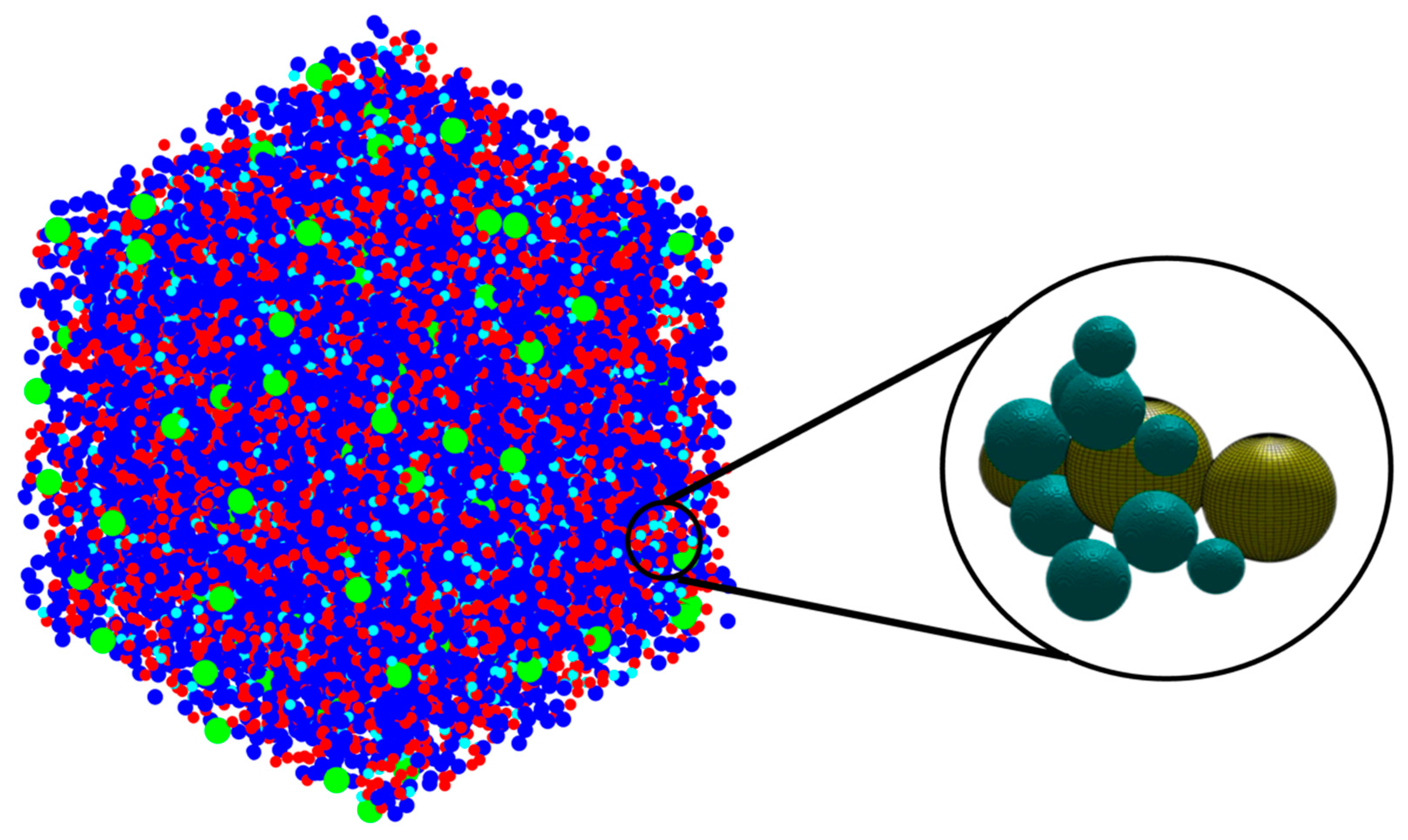
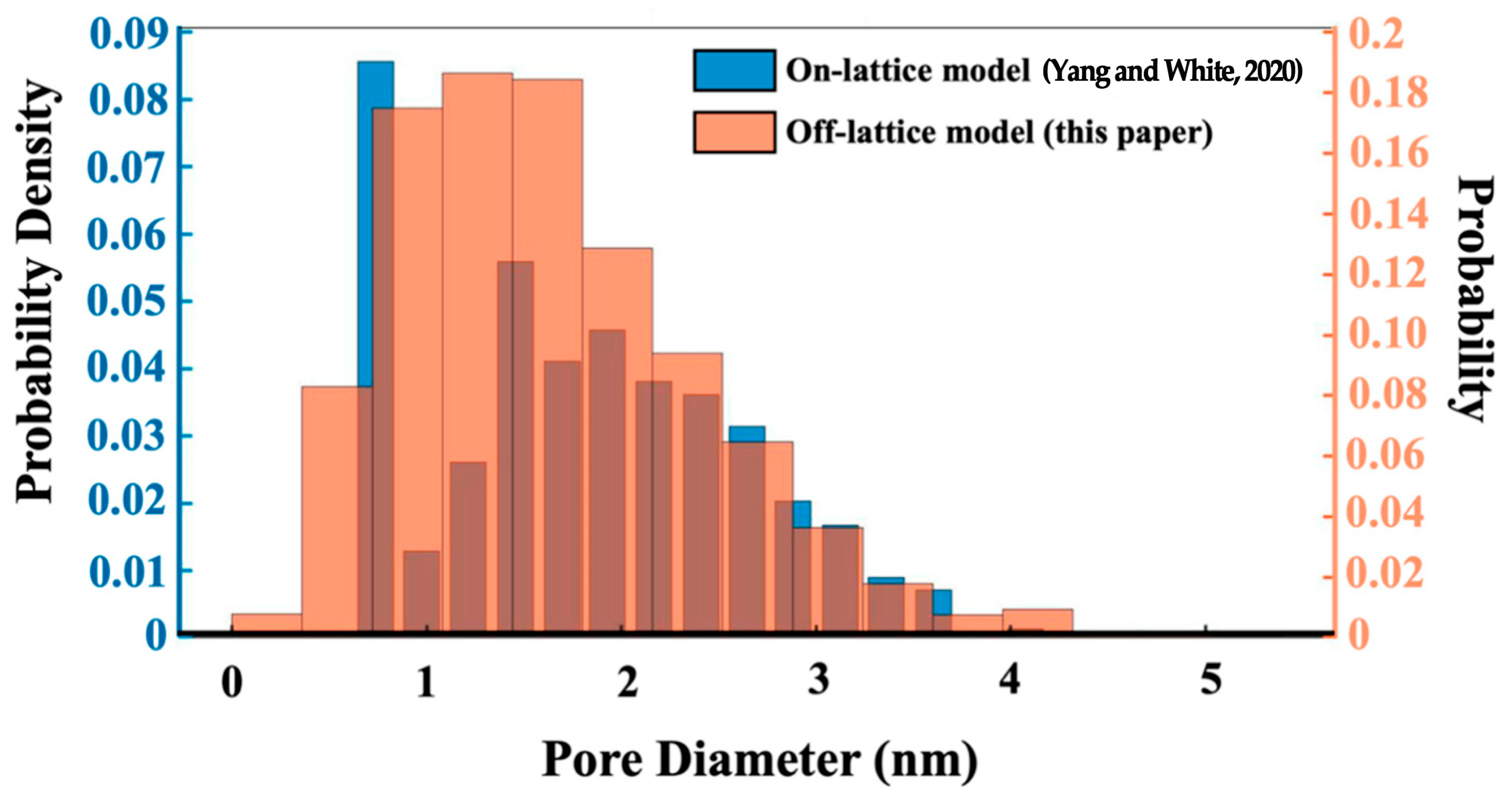
- After the program finished, the global scan method was used to output the cluster size distribution. Next, a clarified pore network model was generated by assuming pores to be spherical. Then, a watershed algorithm and city-block distance transform function were used for digitizing the particle structure, and throats and pore size distribution were deduced.
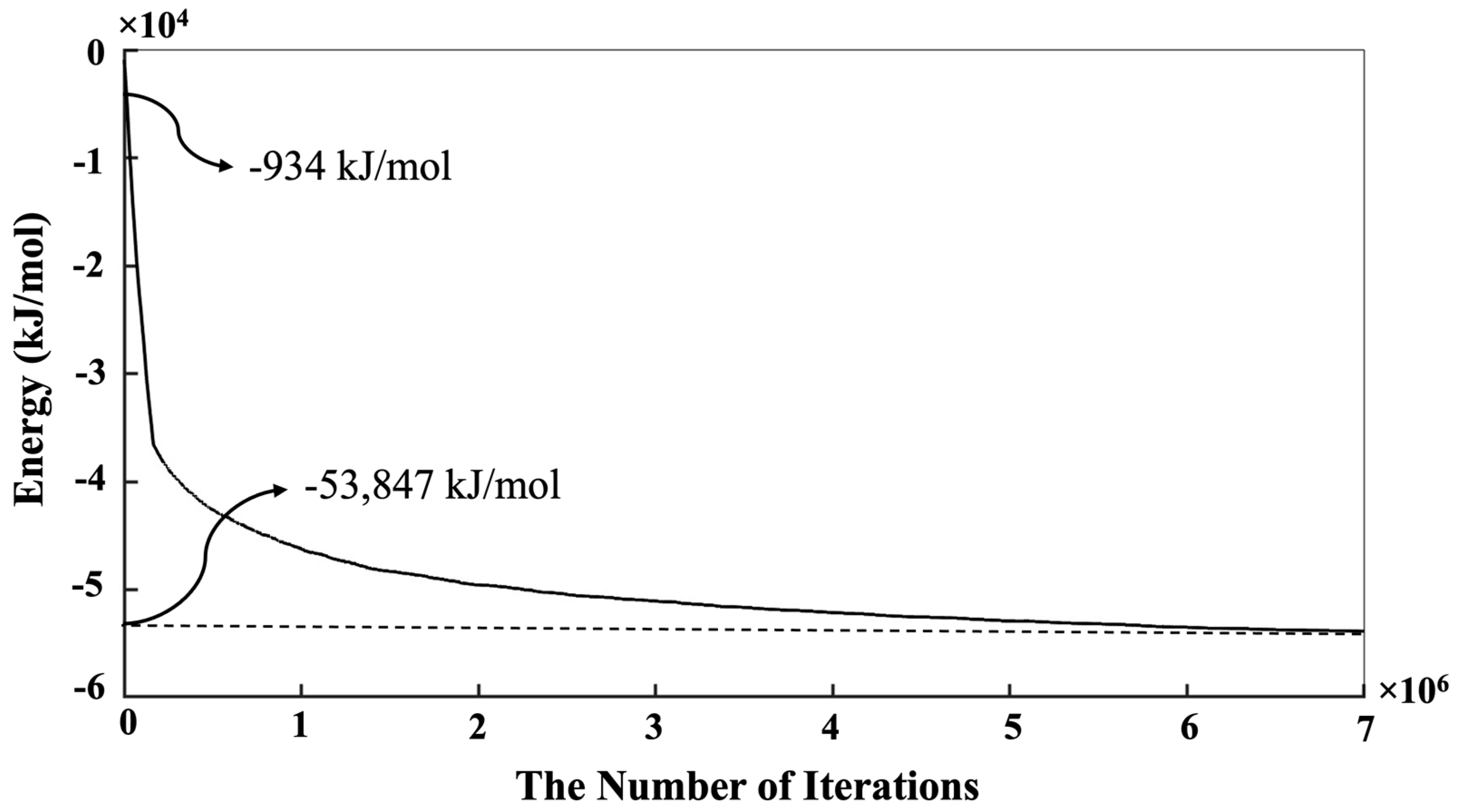
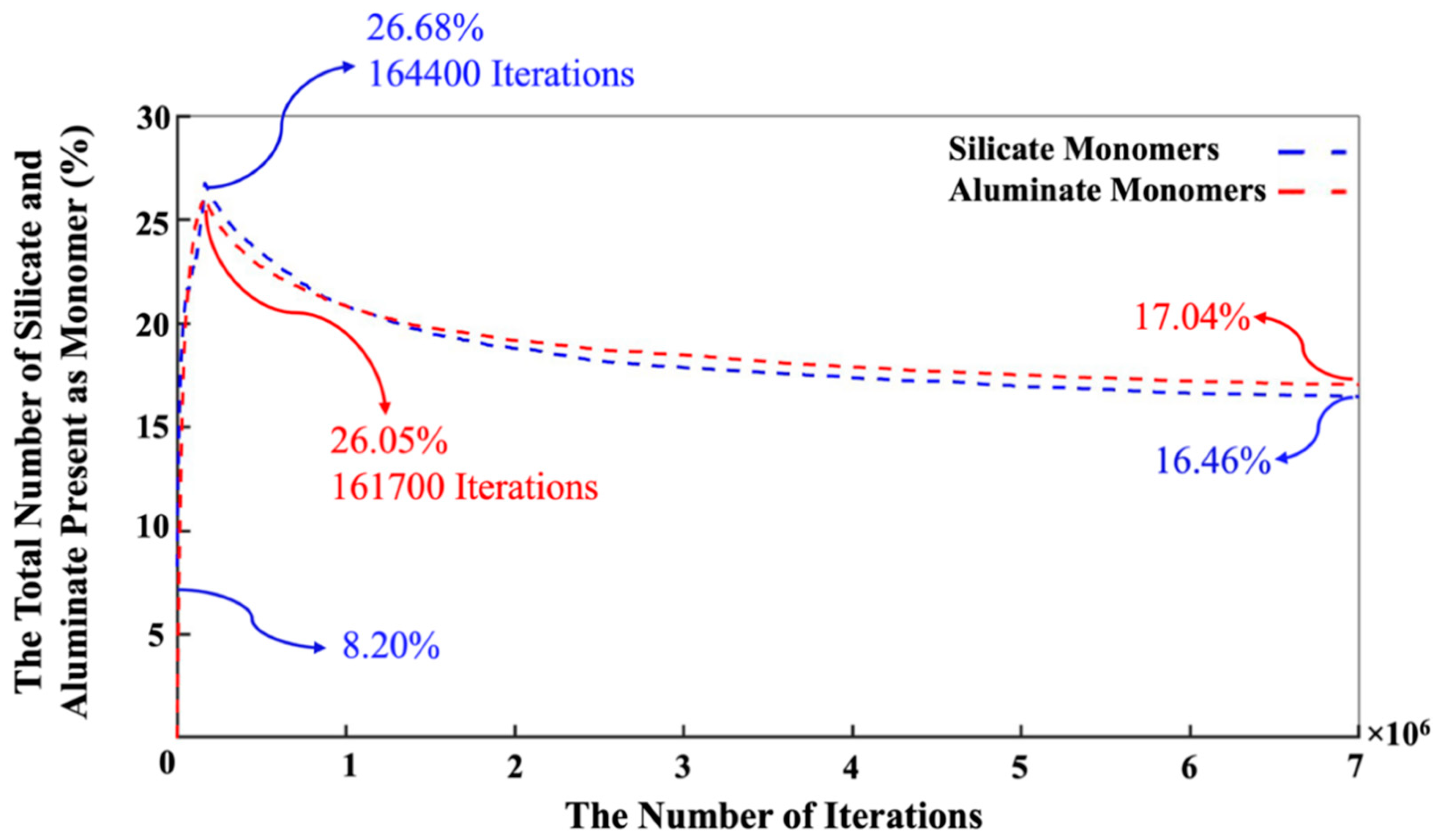
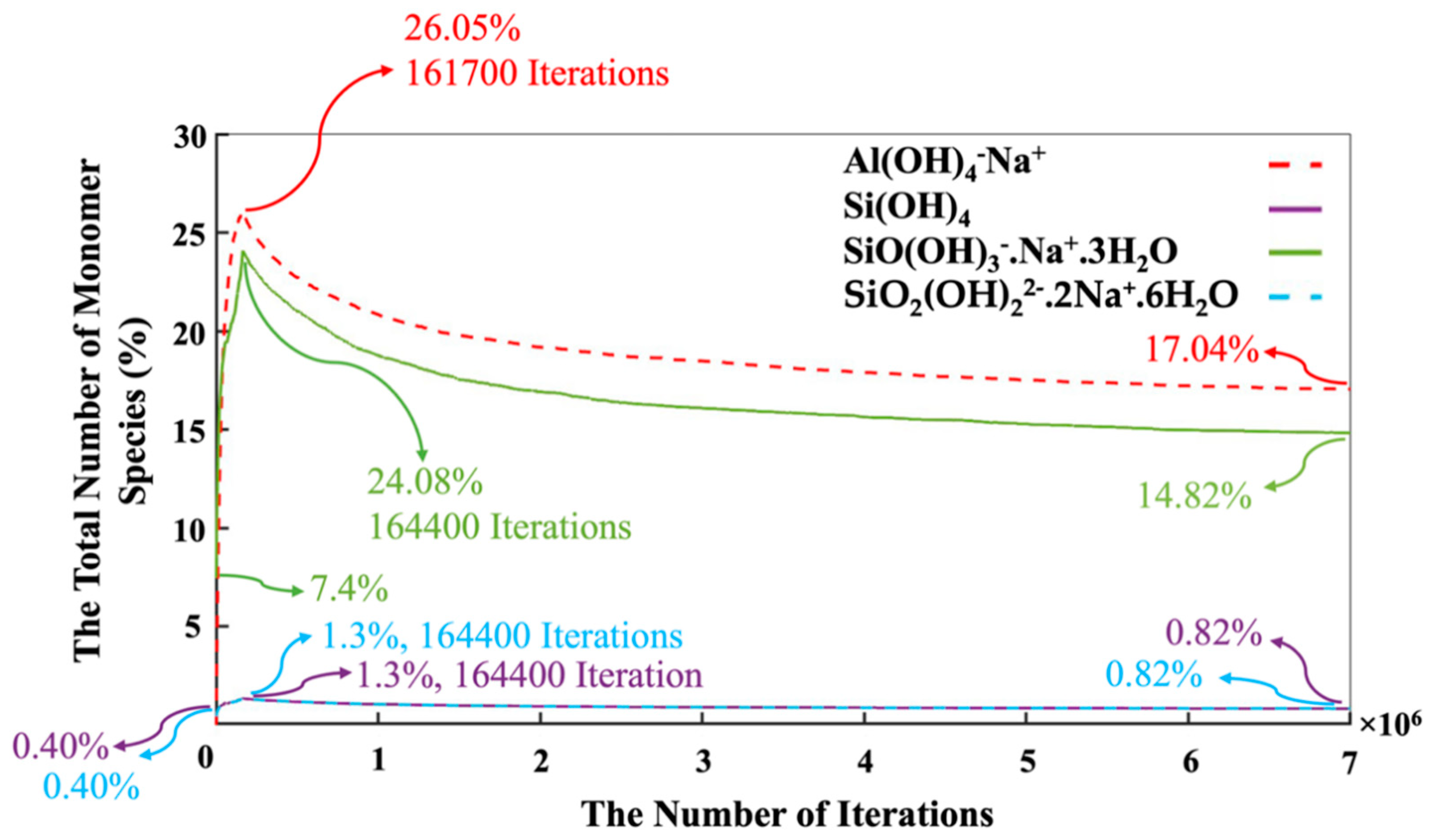
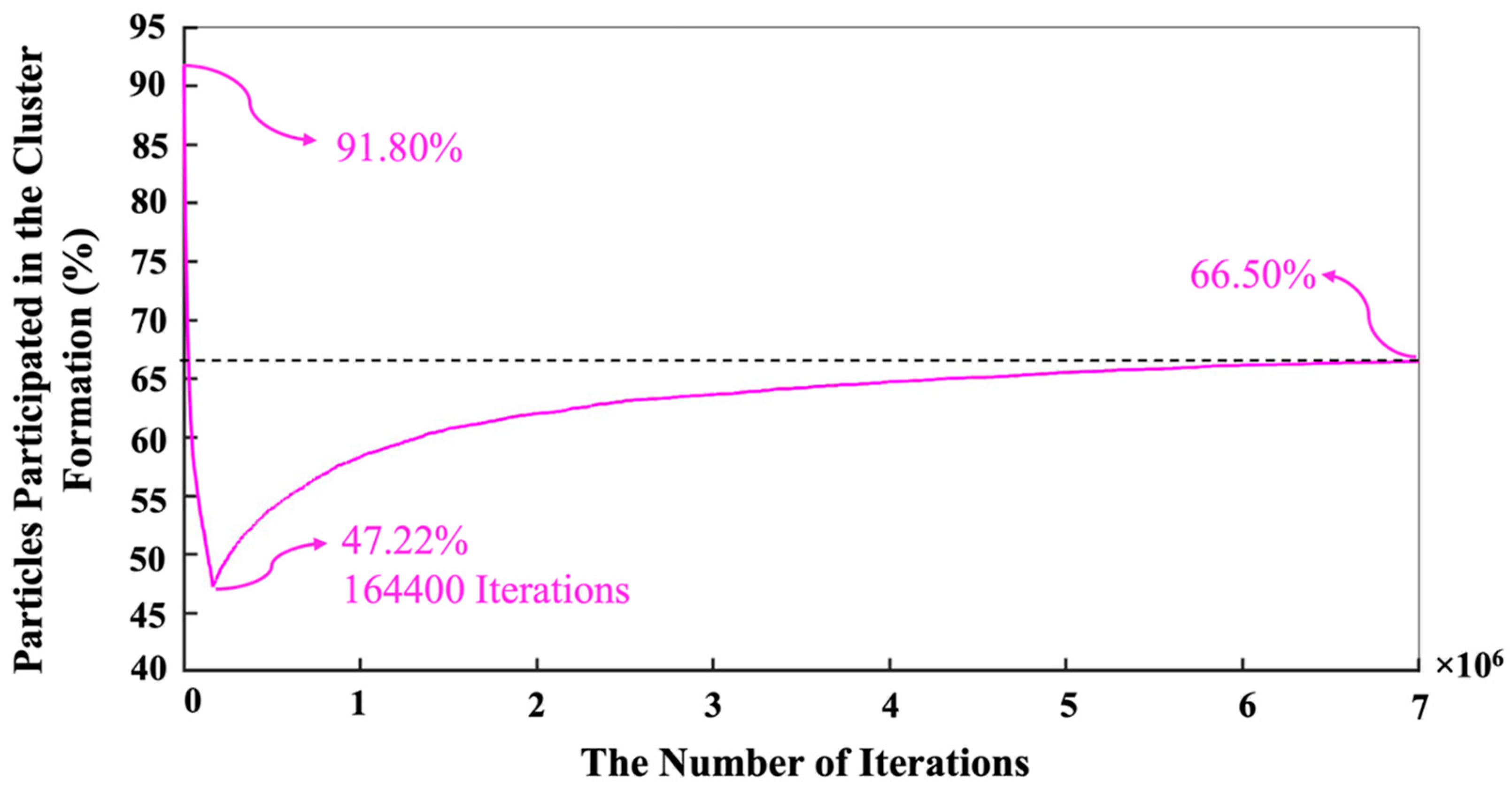
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Pernica, D.; Reis, P.N.B.; Ferreira, J.A.M.; Louda, P. Effect of test conditions on the bending strength of a geopolymer-reinforced composite. J. Mater. Sci. 2010, 45, 744–749. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Steudel, A.; Kleeberg, R.; Kaufhold, S.; Kaltenbach, J.; Schuhmann, R.; Dehn, F.; Emmerich, K. Comprehensive examination of dehydroxylation of kaolinite, disordered kaolinite, and dickite: Experimental studies and density functional theory. Clays Clay Miner. 2020, 68, 319–333. [Google Scholar] [CrossRef]
- Chitsaz, S.; Tarighat, A. Molecular dynamics simulation of N-A-S-H geopolymer macro molecule model for prediction of its modulus of elasticity. Constr. Build. Mater. 2020, 243, 118176. [Google Scholar] [CrossRef]
- Duxson, P.; Fernández-Jiménez, A.; Provis, J.L.; Lukey, G.C.; Palomo, A.; van Deventer, J.S.J. Geopolymer technology: The current state of the art. J. Mater. Sci. 2007, 42, 2917–2933. [Google Scholar] [CrossRef]
- Vogt, O.; Ballschmiede, C.; Ukrainczyk, N.; Koenders, E. Evaluation of Sulfuric Acid-Induced Degradation of Potassium Silicate Activated Metakaolin Geopolymers by Semi-Quantitative SEM-EDX Analysis. Materials 2020, 13, 4522. [Google Scholar] [CrossRef]
- Buchwald, A.; Zellmann, H.-D.; Kaps, C. Condensation of aluminosilicate gels—Model system for geopolymer binders. J. Non-Cryst. Solids 2011, 357, 1376–1382. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Y.; Lin, W.; Liu, Z. In situ monitoring of the hydration process of K-PS geopolymer cement with ESEM. Cem. Concr. Res. 2004, 34, 935–940. [Google Scholar] [CrossRef]
- Vogt, O.; Ukrainczyk, N.; Ballschmiede, C.; Koenders, E. Reactivity and Microstructure of Metakaolin Based Geopolymers: Effect of Fly Ash and Liquid/Solid Contents. Materials 2019, 12, 3485. [Google Scholar] [CrossRef] [Green Version]
- White, C.E.; Provis, J.L.; Proffen, T.; van Deventer, J.S.J. Molecular mechanisms responsible for the structural changes occurring during geopolymerization: Multiscale simulation. AIChE J. 2012, 58, 2241–2253. [Google Scholar] [CrossRef]
- Bakharev, T. Resistance of geopolymer materials to acid attack. Cem. Concr. Res. 2005, 35, 658–670. [Google Scholar] [CrossRef]
- Ukrainczyk, N.; Muthu, M.; Vogt, O.; Koenders, E. Geopolymer, Calcium Aluminate, and Portland Cement-Based Mortars: Comparing Degradation Using Acetic Acid. Materials 2019, 12, 3115. [Google Scholar] [CrossRef] [Green Version]
- Koenig, A.; Herrmann, A.; Overmann, S.; Dehn, F. Resistance of alkali-activated binders to organic acid attack: Assessment of evaluation criteria and damage mechanisms. Constr. Build. Mater. 2017, 151, 405–413. [Google Scholar] [CrossRef]
- Drugă, B.; Ukrainczyk, N.; Weise, K.; Koenders, E.; Lackner, S. Interaction between wastewater microorganisms and geopolymer or cementitious materials: Biofilm characterization and deterioration characteristics of mortars. Int. Biodeterior. Biodegrad. 2018, 134, 58–67. [Google Scholar] [CrossRef]
- Okoye, F.N.; Prakash, S.; Singh, N.B. Durability of fly ash based geopolymer concrete in the presence of silica fume. J. Clean. Prod. 2017, 149, 1062–1067. [Google Scholar] [CrossRef]
- Mortazavi, B.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Zhuang, X.; Shapeev, A.V. Machine-learning interatomic potentials enable first-principles multiscale modeling of lattice thermal conductivity in graphene/borophene heterostructures. Mater. Horiz. 2020, 7, 2359–2367. [Google Scholar] [CrossRef]
- Mortazavi, B.; Yang, H.; Mohebbi, F.; Cuniberti, G.; Rabczuk, T. Graphene or h-BN paraffin composite structures for the thermal management of Li-ion batteries: A multiscale investigation. Appl. Energy 2017, 202, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.-J.; Tse, E.C.M.; Shang, C.; Guo, Z. Nucleation and growth in solution synthesis of nanostructures—From fundamentals to advanced applications. Prog. Mater. Sci. 2022, 123, 100821. [Google Scholar] [CrossRef]
- Harding, J.H.; Freeman, C.L.; Duffy, D.M. Oriented crystal growth on organic monolayers. CrystEngComm 2014, 16, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; White, C.E. Multiscale pore structure determination of cement paste via simulation and experiment: The case of alkali-activated metakaolin. Cem. Concr. Res. 2020, 137, 106212. [Google Scholar] [CrossRef]
- Yang, K.; White, C.E. Modeling the Formation of Alkali Aluminosilicate Gels at the Mesoscale Using Coarse-Grained Monte Carlo. Langmuir 2016, 32, 11580–11590. [Google Scholar] [CrossRef]
- White, C.E.; Provis, J.L.; Proffen, T.; van Deventer, J.S.J. Quantitative Mechanistic Modeling of Silica Solubility and Precipitation during the Initial Period of Zeolite Synthesis. J. Phys. Chem. C 2011, 115, 9879–9888. [Google Scholar] [CrossRef]
- Izadifar, M.; Natzeck, C.; Emmerich, K.; Weidler, P.G.; Gohari, S.; Burvill, C.; Thissen, P. Unexpected Chemical Activity of a Mineral Surface: The Role of Crystal Water in Tobermorite. J. Phys. Chem. C 2022, 126, 12405–12412. [Google Scholar] [CrossRef]
- Izadifar, M.; Königer, F.; Gerdes, A.; Wöll, C.; Thissen, P. Correlation between Composition and Mechanical Properties of Calcium Silicate Hydrates Identified by Infrared Spectroscopy and Density Functional Theory. J. Phys. Chem. C 2019, 123, 10868–10873. [Google Scholar] [CrossRef]
- Izadifar, M.; Dolado, J.S.; Thissen, P.; Ayuela, A. Interactions between Reduced Graphene Oxide with Monomers of (Calcium) Silicate Hydrates: A First-Principles Study. Nanomaterials 2021, 11, 2248. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of Portlandite in Pure Water: Part 1 Molecular Dynamics (MD) Approach. Materials 2022, 15, 1404. [Google Scholar] [CrossRef]
- Salah Uddin, K.M.; Izadifar, M.; Ukrainczyk, N.; Koenders, E.; Middendorf, B. Dissolution of β-C2S Cement Clinker: Part 1 Molecular Dynamics (MD) Approach for Different Crystal Facets. Materials 2022, 15, 6388. [Google Scholar] [CrossRef]
- Izadifar, M.; Thissen, P.; Abadi, R.; Jam, A.N.; Gohari, S.; Burvill, C.; Rabczuk, T. Fracture toughness of various percentage of doping of boron atoms on the mechanical properties of polycrystalline graphene: A molecular dynamics study. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 114, 113614. [Google Scholar] [CrossRef]
- Jam, A.N.; Abadi, R.; Izadifar, M.; Rabczuk, T. Molecular dynamics study on the mechanical properties of carbon doped single-layer polycrystalline boron-nitride nanosheets. Comput. Mater. Sci. 2018, 153, 16–27. [Google Scholar] [CrossRef]
- Rahier, H.; Wastiels, J.; Biesemans, M.; Willlem, R.; Van Assche, G.; Van Mele, B. Reaction mechanism, kinetics and high temperature transformations of geopolymers. J. Mater. Sci. 2007, 42, 2982–2996. [Google Scholar] [CrossRef]
- Šefčík, J.; Rankin, S.E. Monte Carlo Simulations of Size and Structure of Gel Precursors in Silica Polycondensation. J. Phys. Chem. B 2003, 107, 52–60. [Google Scholar] [CrossRef]
- Katsoulakis, M.A.; Vlachos, D.G. Coarse-grained stochastic processes and kinetic Monte Carlo simulators for the diffusion of interacting particles. J. Chem. Phys. 2003, 119, 9412–9427. [Google Scholar] [CrossRef]
- Dong, H.; Blunt, M.J. Pore-network extraction from micro-computerized-tomography images. Phys. Rev. E 2009, 80, 036307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, A.; Jamshidi, S.; Salehi, S. An automated simple algorithm for realistic pore network extraction from micro-tomography images. J. Pet. Sci. Eng. 2014, 123, 164–171. [Google Scholar] [CrossRef]
- Šefčík, J.; McCormick, A.V. Thermochemistry of aqueous silicate solution precursors to ceramics. AIChE J. 1997, 43, 2773–2784. [Google Scholar] [CrossRef]
- Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.M.; Middendorf, B.; Koenders, E. Dissolution of Portlandite in Pure Water: Part 2 Atomistic Kinetic Monte Carlo (KMC) Approach. Materials 2022, 15, 6716. [Google Scholar] [CrossRef]
- Izadifar, M.; Ukrainczyk, N.; Salah Uddin, K.; Middendorf, B.; Koenders, E. Dissolution of β-C2S Cement Clinker: Part 2 Atomistic Kinetic Monte Carlo (KMC) Upscaling Approach. Materials 2022, 15, 1442. [Google Scholar] [CrossRef]
- Hul, G.; Seijo, M.; Carnal, F.; Cornelis, G.; Stoll, S. Effect of deposition, detachment and aggregation processes on nanoparticle transport in porous media using Monte Carlo simulations. Environ. Sci. Nano 2021, 8, 2223–2234. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
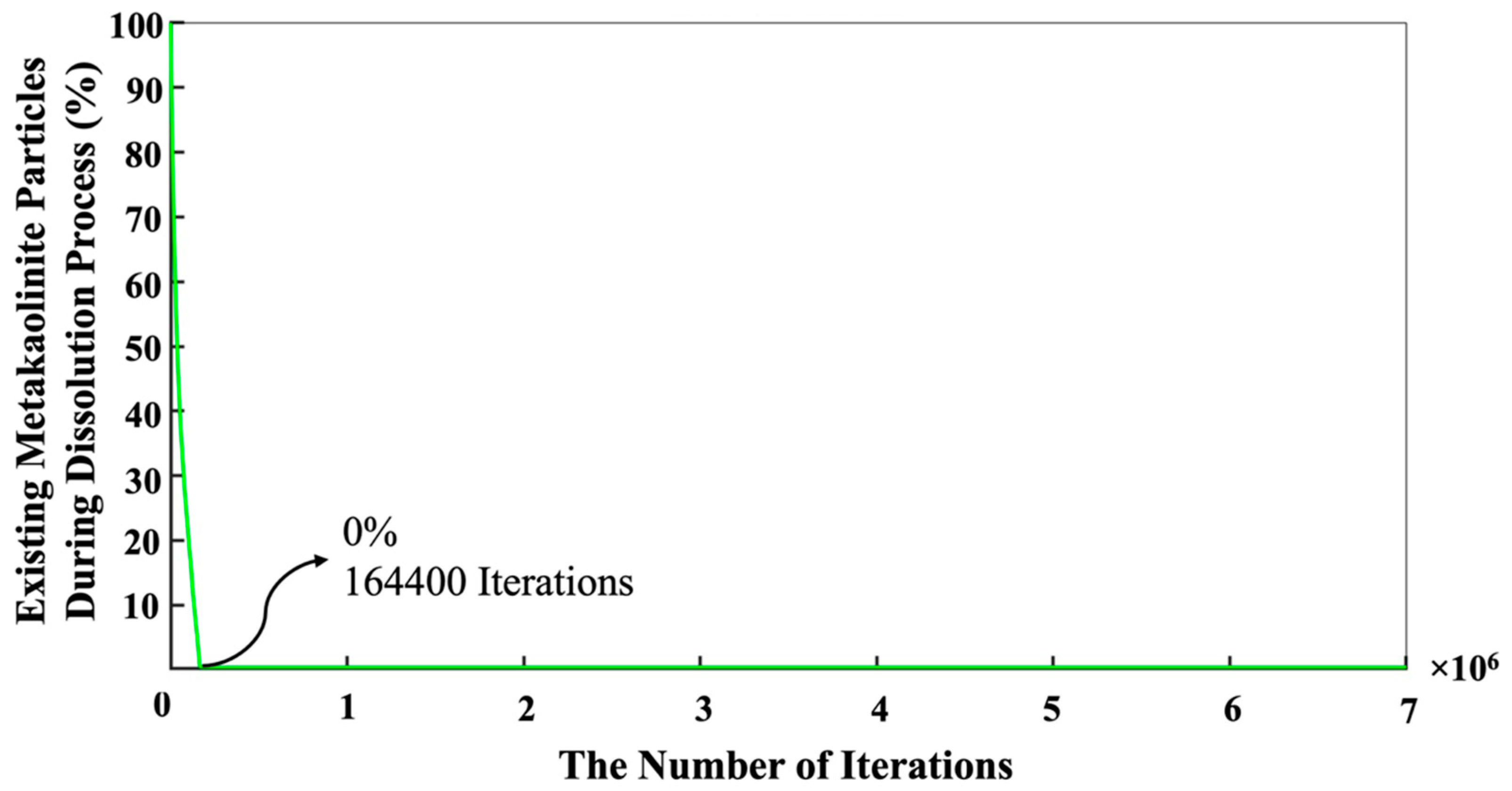{kind=link}
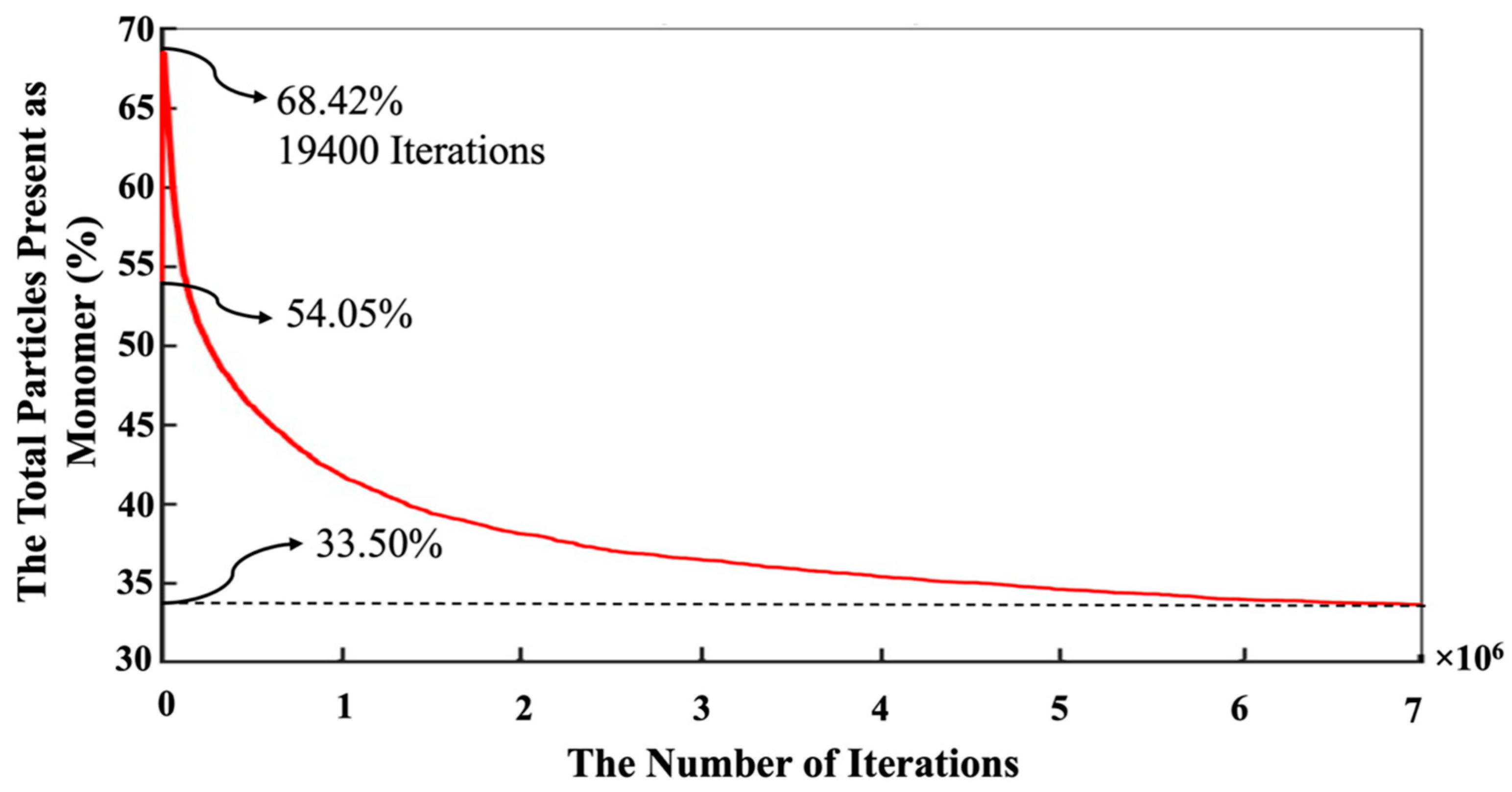{kind=link}
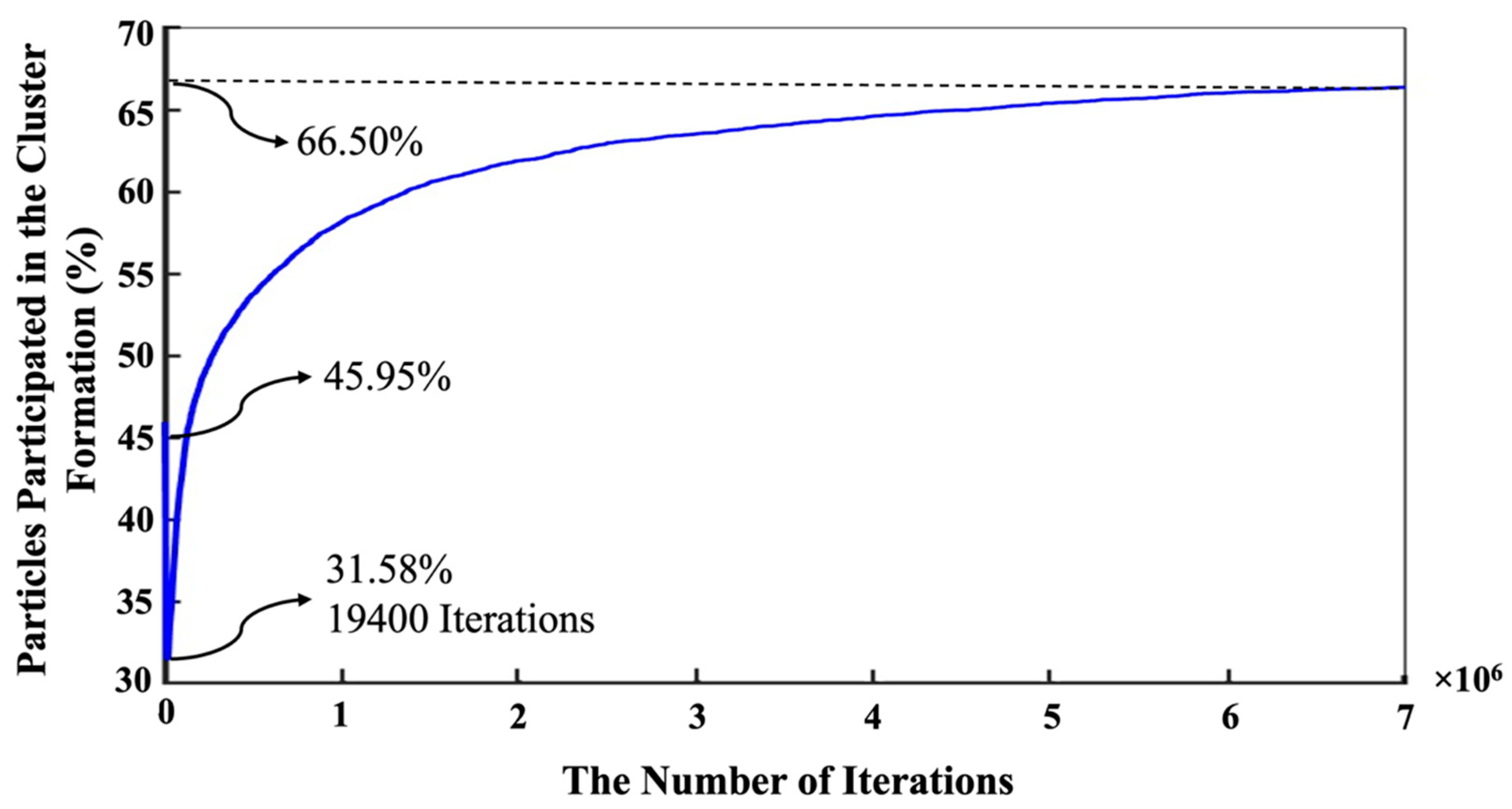{kind=link}
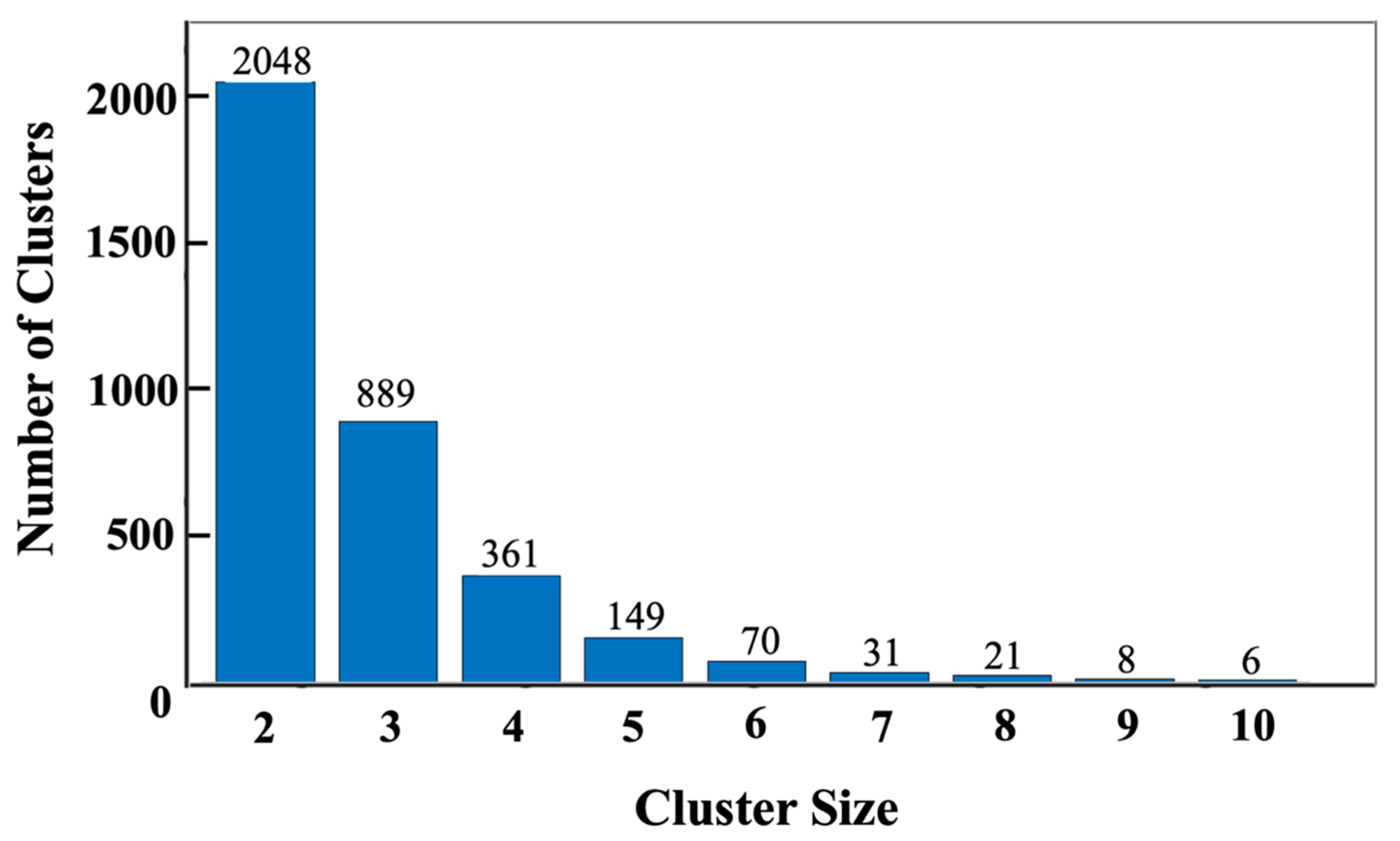{kind=link}
{kind=link}
{kind=link}
| Monomer Species | M | M−·Na·3H2O | M2−·2Na·6H2O | A−·Na |
|---|---|---|---|---|
| M | −1.8 | −9.3 | −5.3 | −21.2 |
| M−·Na·3H2O | −0.9 | 8.1 | −9.7 | |
| M2−·2Na·6H2O | 35.0 | 14.5 | ||
| A−·Na | 16.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izadifar, M.; Valencia, N.C.; Xiao, P.; Ukrainczyk, N.; Koenders, E. 3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels. Materials 2023, 16, 1863. https://doi.org/10.3390/ma16051863
Izadifar M, Valencia NC, Xiao P, Ukrainczyk N, Koenders E. 3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels. Materials. 2023; 16(5):1863. https://doi.org/10.3390/ma16051863
Chicago/Turabian StyleIzadifar, Mohammadreza, Nicolas Castrillon Valencia, Peng Xiao, Neven Ukrainczyk, and Eduardus Koenders. 2023. "3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels" Materials 16, no. 5: 1863. https://doi.org/10.3390/ma16051863
APA StyleIzadifar, M., Valencia, N. C., Xiao, P., Ukrainczyk, N., & Koenders, E. (2023). 3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels. Materials, 16(5), 1863. https://doi.org/10.3390/ma16051863