Efficient Syntheses of [(n-C4H9)4N]4[α-Mo8O26] and [(n-C4H9)4N]2[Mo2O7]

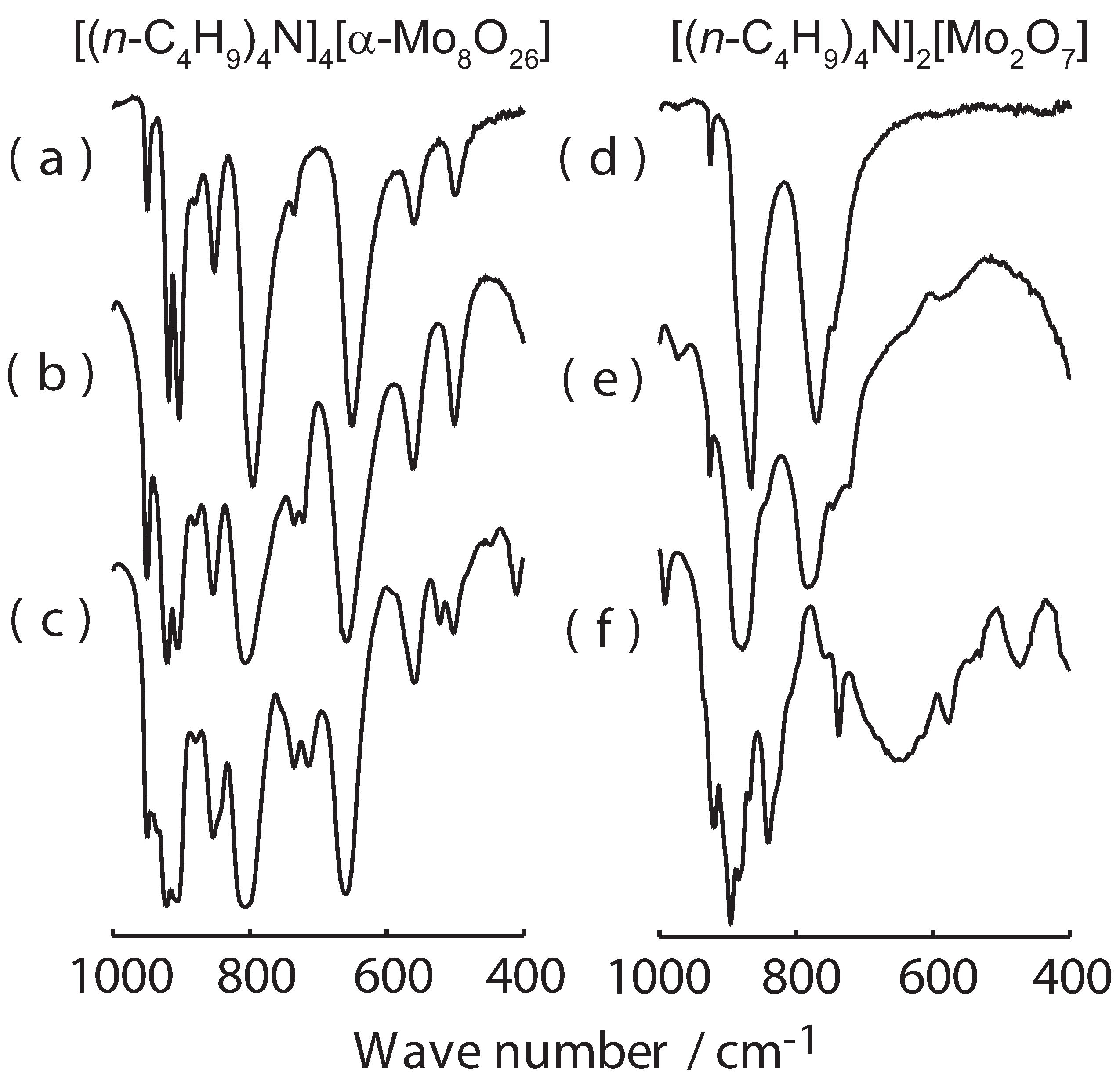{kind=link}
Abstract
:1. Introduction
2. Results and Discussion

3. Experimental
3.1. General
4. Conclusions
References and Notes
- Fuchs, J.; Hartl, H. Structure of tetrabutylammonium octamolybdate. Angew. Chem. Int. Ed. Engl. 1976, 15, 375–376. [Google Scholar] [CrossRef]
- Klemperer, W.G.; Shum, W. Synthesis and interconversion of the isomeric α- and β-Mo8O264- ions. J. Am. Chem. Soc. 1976, 98, 8291–8293. [Google Scholar] [CrossRef]
- Day, V.W.; Fredrcih, M.F.; Klemperer, W.G.; Shum, W. Structural and dynamic stereochemistry of α-Mo8O264-. J. Am. Chem. Soc. 1977, 99, 952–953. [Google Scholar] [CrossRef]
- Day, V.W.; Fredrcih, M.F.; Klemperer, W.G.; Shum, W. Synthesis and characterization of the dimolybdate ion, Mo2O72-. J. Am. Chem. Soc. 1977, 99, 6146–6148. [Google Scholar] [CrossRef]
- Nugent, W.A. In situ-generated molybdenum(VI) reagent for cis-chlorination of alkenes. Tetrahedron Lett. 1978, 37, 3427–3430. [Google Scholar] [CrossRef]
- Day, V.W.; Fredrich, M.F.; Klemperer, W.G.; Liu, R.S. Polyxomolybdate-hydrocarbon interactions. Synthesis and structure of the CH2Mo4O15H3- anion and related methylene-dioxymolybdates. J. Am. Chem. Soc. 1979, 101, 491–492. [Google Scholar]
- Do, Y.; Simhon, E.D.; Holm, R.H. Oxo/Sulfido ligand substitutuion in [Mo2O7]2-: Reaction sequence and characterization of the final product, [MoS3(OSiM3)]-. Inorg. Chem. 1985, 24, 1831–1838. [Google Scholar] [CrossRef]
- Che, T.M.; Day, V.W.; Francesconi, L.C.; Fredrich, M.F.; Klemperer, W.G.; Shum, W. Synthesis and structure of the [CpTi(Mo5O18)]3- and [CpTi(W5O18)]3- anions. Inorg. Chem. 1985, 24, 4055–4062. [Google Scholar] [CrossRef]
- Liu, S.; Shaikh, S.N.; Zubieta, J. Polyoxomolybdate-o-benzoquinone interactions. Synthesis and structure of a diacetal derivative, [Mo4O15(OH)(C14H8)]3-, from 9,10-phenanthrenequinone carbonyl insertion. Comparison to the reaction products with tetrachloro-1,2-benzoquinone, the ligand-bridged binuclear complexes [(MoO2Cl2)2L]2-, L = (C6Cl2O4)2- and (C2O4)2-, formed via carbonyl insertion and chloride transfer. Inorg. Chem. 1988, 27, 3064–3066. [Google Scholar]
- Chen, Q.; Liu, S.; Zhu, H.; Zubieta, J. Carbomolybdate clusters formed by carbonyl insertion into molybdenum-oxygen bonds. Structural investigation of the [RMo4O15X]3- core (R = -C9H4O, X = OCH3; R = -C14H10, X = -C7H5O2; R = C14H8, X = -OH). Polyhedron 1989, 8, 2915–2923. [Google Scholar]
- Ichida, H.; Yagasaki, A. Synthesis and structure of the novel polymolybdate which contains five-coordinated Mo(VI). J. Chem. Soc., Chem. Commun. 1991, 27–28. [Google Scholar]
- Kitamura, A.; Ozeki, T.; Yagasaki, A. β-Octamolybdate as a building block. Synthesis and structural characterization of rare earth–molybdate adducts. Inorg. Chem. 1997, 36, 4275–4279. [Google Scholar]
- Proust, A.; Thouvenot, R.; Herson, P. Revisiting the synthesis of [Mo6Cp*O18]-. X-Ray structural analysis, UV-visible, electrochemical and multinuclear NMR characterization. J. Chem. Soc., Dalton Trans. 1999, 51–55. [Google Scholar]
- Wang, X.-J.; Chen, Z.-F.; Kang, B.-S.; Liang, H.; Liu, H.-Q.; Yu, K.-B.; Su, C.-Y.; Chen, Z.-N. Chemistry of 2-mercaptophenol (H2mp). Part iii. One dimensional arrays of binuclear anions of Mo (VI) linked by bis (2-hydroxyphenyl) disulfide via hydrogen bondings. Polyhedron 1999, 18, 647–655. [Google Scholar]
- Ozeki, T.; Sakaguchi, M.; Yagasaki, A. Synthesis and structure of a novel dimeric molybdenum oxyhalide anion. Polyhedron 2000, 19, 43–45. [Google Scholar] [CrossRef]
- Nishikawa, K.; Kinoshita, I.; Nishioka, T.; Isobe, K. Synthesis and X-ray analysis of Mn-Mo mixed metal oxide cluster with cubic framework. Chem. Lett. 2000, 1342–1343. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, M.; Cheung, C.F.; Barnes, C.L.; Peng, Z. Functionalization of [MoW5O19]2- with aromatic amines: Synthesis of the first arylimido derivatives of mixed-metal polyoxometalates. Inorg. Chem. 2001, 40, 5489–5490. [Google Scholar] [CrossRef] [PubMed]
- Hasenknopf, B.; Delmont, R.; Herson, P.; Gouzerh, P. Anderson-type heteropolymolybdates containing tris(alkoxo) ligands: Synthesis and structural characterization. Eur. J. Inorg. Chem. 2002, 1081–1087. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Z.-F.; Hu, R.-X.; Yu, Q.; Zhou, Z.-Y.; Zhou, X-G. Synthesis, crystal structure and spectroscopic characterization of mono- and tetra-nuclear molybdenum(VI) complexes with 2-mercaptopyridine N-oxide: (n-Bu4N)2[MoO2(PT)2·Mo4O8(μ-O)2(μ3-O)2(PT)2]. Trans. Met. Chem. 2002, 27, 102–104. [Google Scholar]
- Marcoux, P.R.; Hasenknopf, B.; Vaissermann, J.; Gouzerh, P. Developing remote metal binding sites in heteropolymolybdates. Eur. J. Inorg. Chem. 2003, 2406–2412. [Google Scholar] [CrossRef]
- Bustos, C.; Hasenknopf, B.; Thouvenot, R.; Vaissermann, J.; Proust, A.; Gouzerh, P. Lindqvist-type (aryldiazenido)polyoxomolybdates - synthesis, and structural and spectroscopic characterization of compounds of the type (nBu4N)3[Mo6O18(N2Ar)]. Eur. J. Inorg. Chem. 2003, 2757–2766. [Google Scholar] [CrossRef]
- Roh, S.-G.; Proust, A.; Robert, F.; Gouzerh, P. Coordination chemistry of amidoximes. Molybdenum complexes of (alkyl)aminoacetamidoximes and fumaromonoamidoxime. Inorg. Chim. Acta 2003, 342, 311–315. [Google Scholar] [CrossRef]
- Laurencin, D.; Fidalgo, E.G.; Villanneau, R.; Villain, R.; Herson, P.; Pacifico, J.; Stoeckli-Evans, H.; Benard, M.; Rohmer, M.-M.; Suess-Fink, G.; Proust, A. Framework fluxionality of organometallic oxides: Synthesis, crystal structure, EXAFS, and DFT studies on [{Ru(η6-arene)}4Mo4O16] complexes. Chem. Eur. J. 2004, 10, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Li, Q.; Ge, N.; Wei, Y.; Wang, Y.; Wang, P.; Guo, H. An easy route to monofunctionalized organoimido derivatives of the lindqvist hexamolybdate. Eur. J. Inorg. Chem. 2004, 2819–2822. [Google Scholar] [CrossRef]
- Li, Q.; Wu, P.; Xia, Y.; Wei, Y.; Guo, Y. Synthesis, spectroscopic studies and crystal structure of a polyoxoanion cluster incorporating para-bromophenylimido ligand, (Bu4N)2[Mo6O18(NC6H4Br-p)]. J. Organomet. Chem. 2006, 691, 1223–1228. [Google Scholar] [CrossRef]
- Qui, Y.; Xu, L.; Gao, G.; Wang, W.; Li, F. A new arylimido derivative of polyoxometalate (Bu4N)2[Mo6O17(NAr)2] [Ar = 2,6-(CH3)2C6H3]: Synthesis, structure and physicochemical properties. Inorg. Chim. Acta 2006, 359, 451–458. [Google Scholar] [CrossRef]
- Xu, L.; Lu, M.; Xu, B.; Wei, Y.; Peng, Z.; Powell, D.R. Towards main-chain-polyoxometalate-containing hybrid polymers: A highly efficient approach to bifunctionalized organoimido derivatives of hexamolybdates. Angew. Chem. Int. Ed. Eng. 2002, 41, 4129–4132. [Google Scholar] [CrossRef]
- Klemperer, W.G. Tetrabutylammonium isopolyoxometalates. Inorg. Syntheses 1990, 27, 74–85. [Google Scholar]
- Compound I does not react with KBr when the latter is thoroughly dried and a spectrum of pure I is obtained. See ref. [2]
- Honda, D.; Ikegami, S.; Inoue, T.; Ozeki, T.; Yagasaki, A. Protonation and methylation of an anderson-type polyoxoanion [IMo6O24]5-. Inorg. Chem. 2007, 46, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ikegami, S.; Yagasaki, A. Efficient Syntheses of [(n-C4H9)4N]4[α-Mo8O26] and [(n-C4H9)4N]2[Mo2O7]. Materials 2009, 2, 869-875. https://doi.org/10.3390/ma2030869
Ikegami S, Yagasaki A. Efficient Syntheses of [(n-C4H9)4N]4[α-Mo8O26] and [(n-C4H9)4N]2[Mo2O7]. Materials. 2009; 2(3):869-875. https://doi.org/10.3390/ma2030869
Chicago/Turabian StyleIkegami, Shusaku, and Atsushi Yagasaki. 2009. "Efficient Syntheses of [(n-C4H9)4N]4[α-Mo8O26] and [(n-C4H9)4N]2[Mo2O7]" Materials 2, no. 3: 869-875. https://doi.org/10.3390/ma2030869
APA StyleIkegami, S., & Yagasaki, A. (2009). Efficient Syntheses of [(n-C4H9)4N]4[α-Mo8O26] and [(n-C4H9)4N]2[Mo2O7]. Materials, 2(3), 869-875. https://doi.org/10.3390/ma2030869