Characterization and Biocompatibility of Biopolyester Nanofibers

Abstract
:1. General Introduction
1.1. Biodegradable polymers for biomedical applications

1.2. Preparation of nanofibers of biodegradable polymers by electrospinning
2. Preparation and Characterization of PHA and PLA Nanofibers
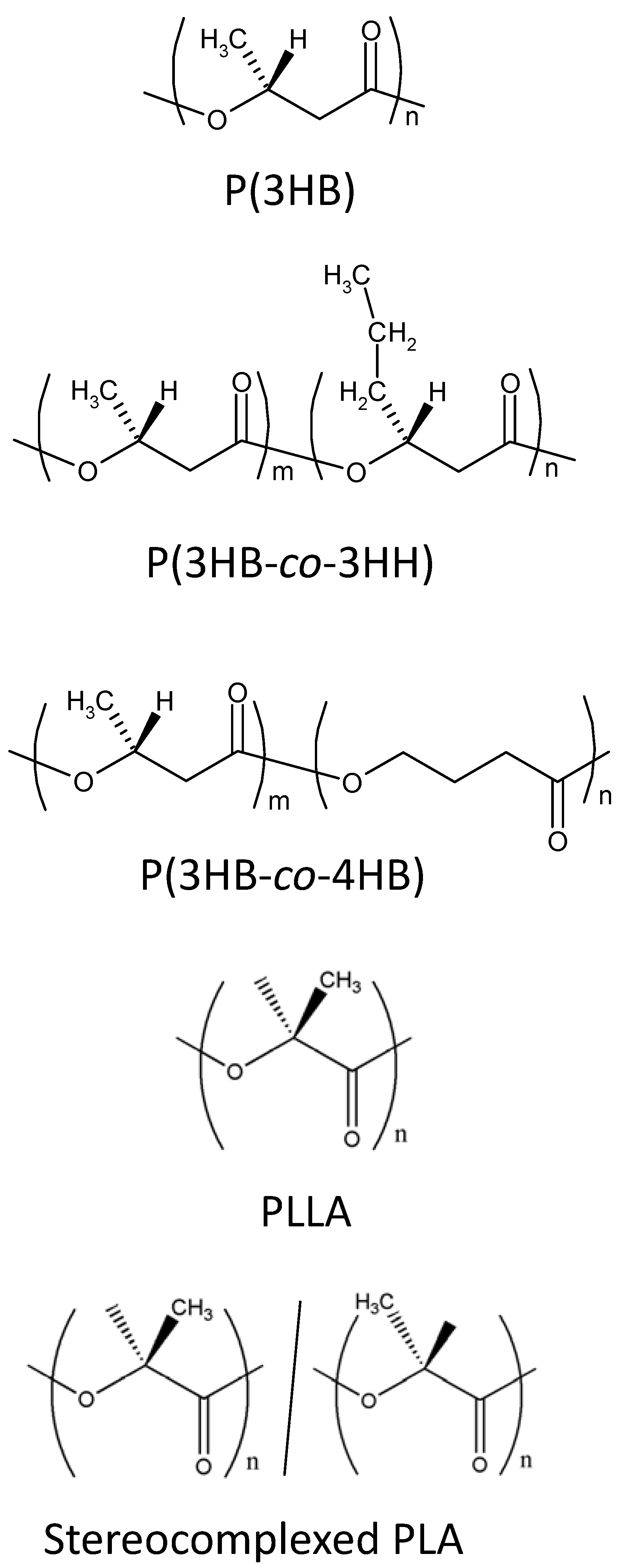
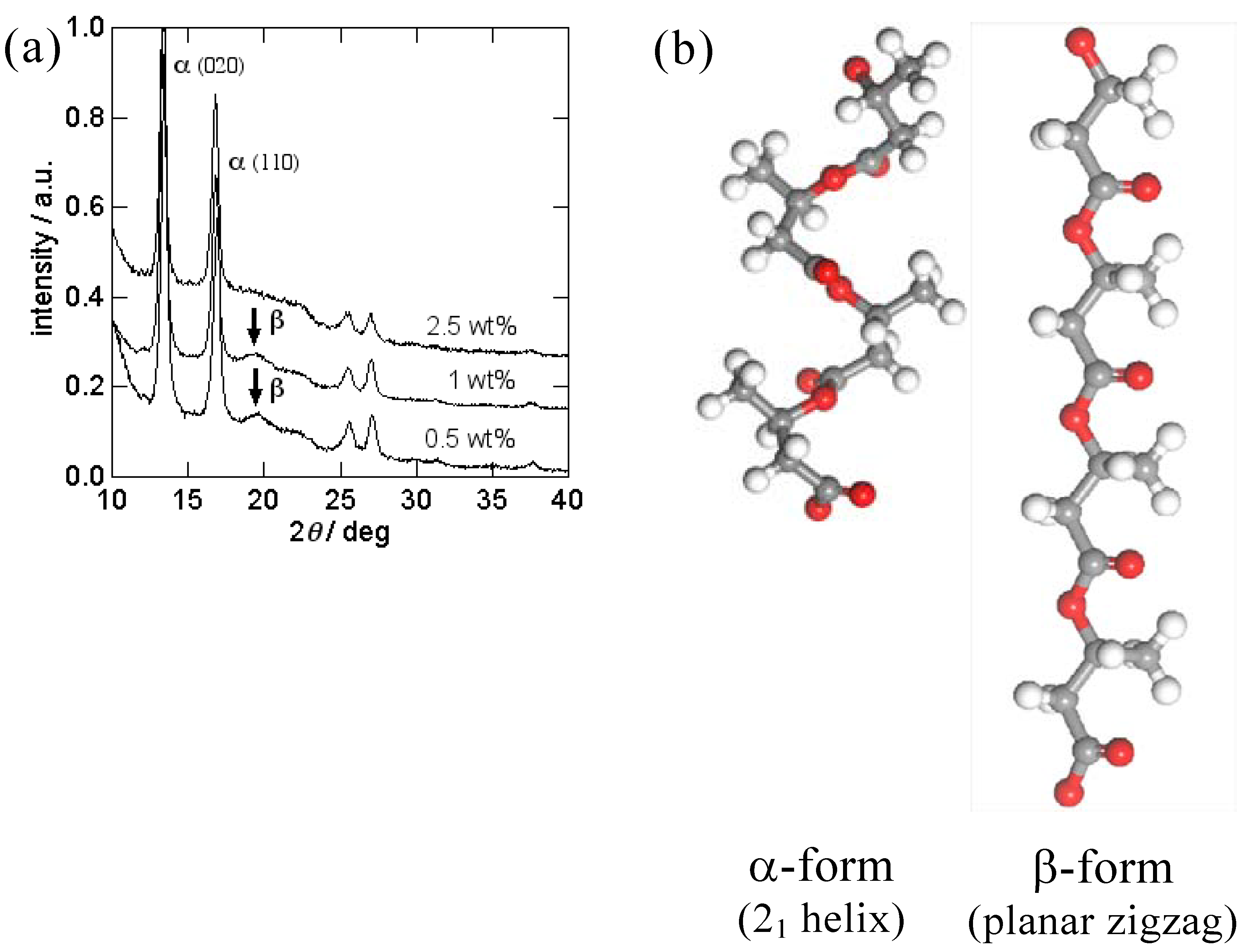
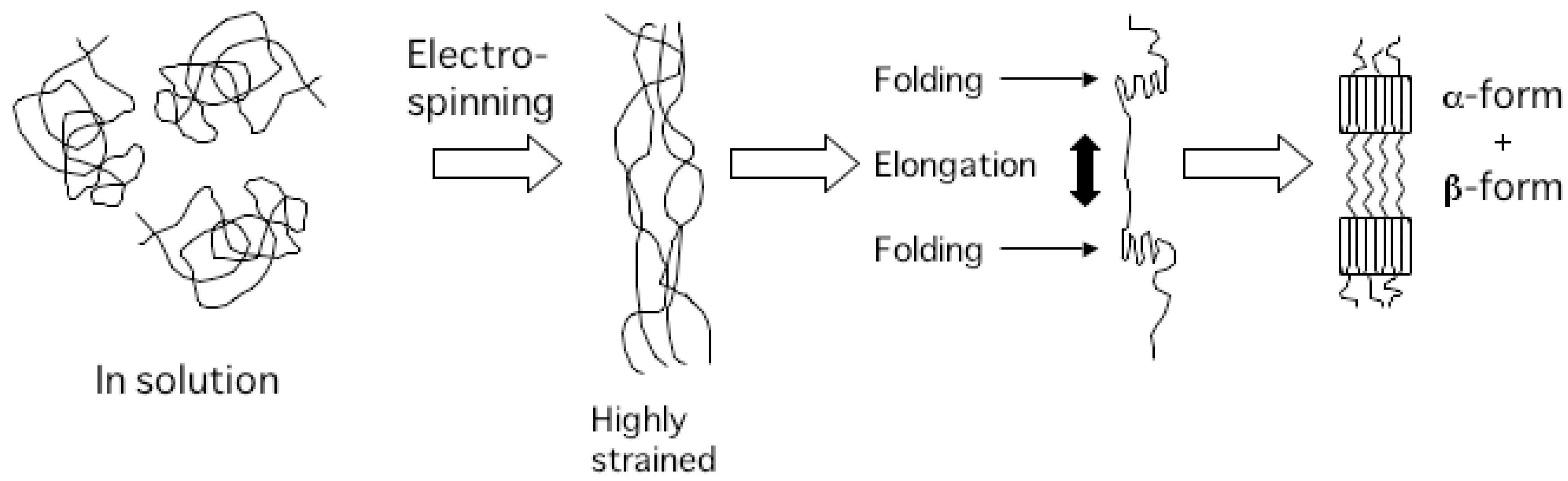
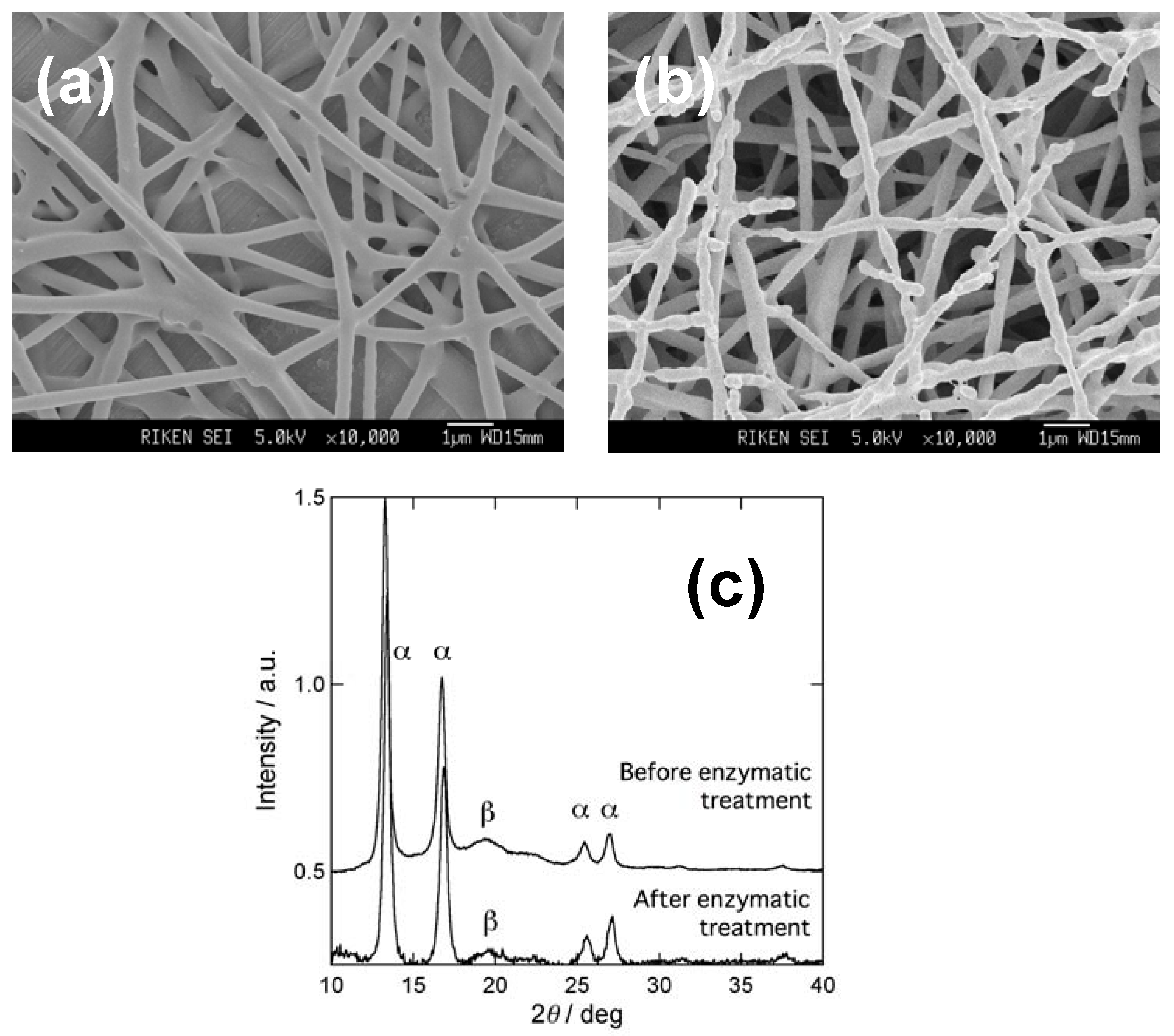
2.1. PHA nanofibers




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Condition | Mechanical properties | |
|---|---|---|---|
| Tensile strength (MPa) | Young’s Modulus (MPa) | ||
| P(3HB) | As-spun | 17 | 223 |
| Sterilized | 15 | 234 | |
| 4 weeks in vivo in vitro | 12 14 | 182 220 | |
| 12 weeks in vivo in vitro | 15 13 | 152 194 | |
| P(3HB-co-5mol%-3HH) | As-spun | 15 | 277 |
| Sterilized | 12 | 272 | |
| 4 weeks in vivo in vitro | 12 13 | 268 208 | |
| 12 weeks in vivo in vitro | ND b 15 | ND b 230 | |
| P(3HB-co-7mol%-4HB) | As-spun | 8 | 184 |
| Sterilized | 8 | 139 | |
| 4 weeks in vivo in vitro | ND b 8 | ND b 163 | |
| 12 weeks in vivo in vitro | ND b 9 | ND b 110 | |
| P(3HB-co-97mol%-4HB) | As-spun | 13 | 9 |
| Sterilized | 15 | 16 | |
| 4 weeks in vivo in vitro | 4 11 | 12 14 | |
| 12 weeks in vivo in vitro | ND b 14 | ND b 16 | |
| Skina | 5-30 | 15-150 | |
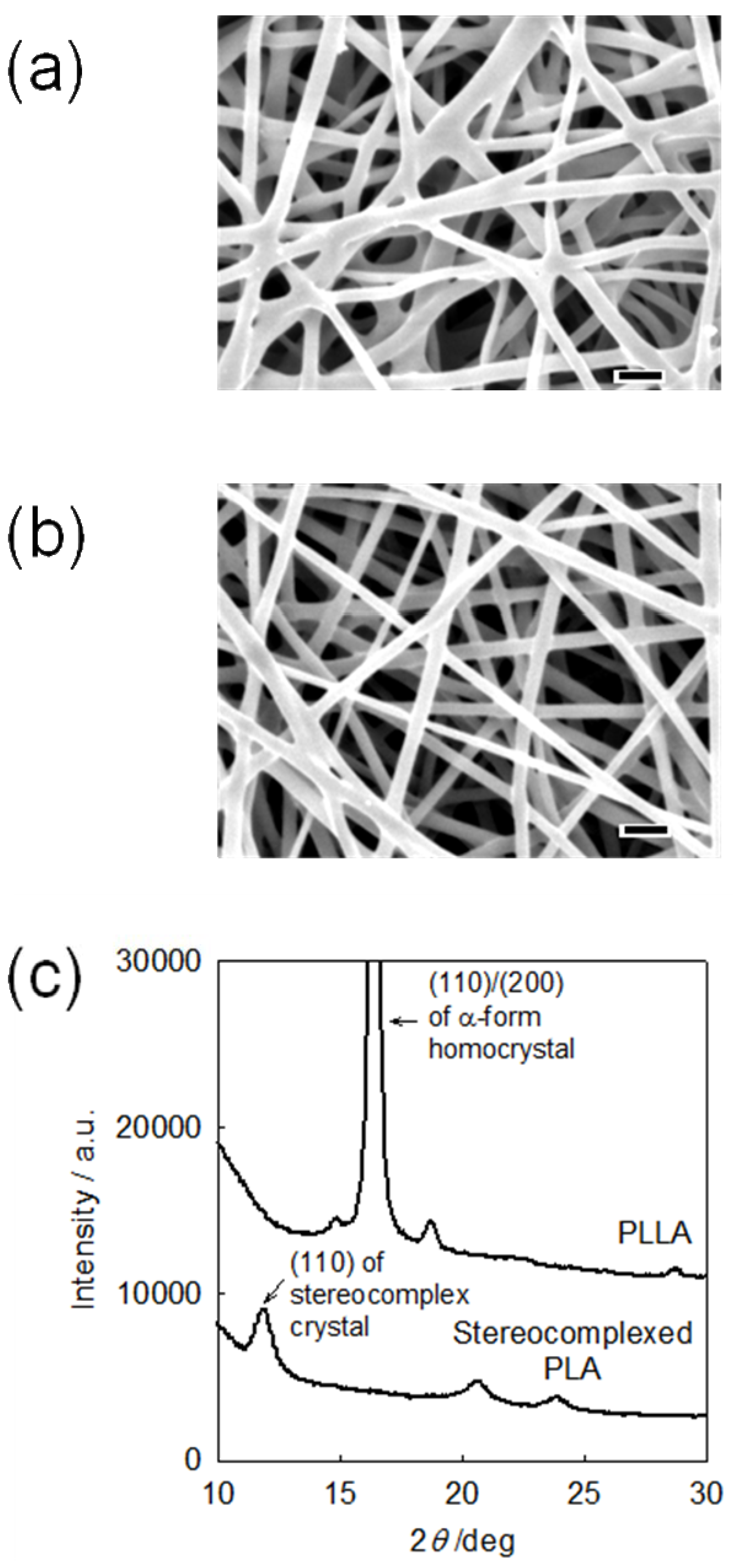
2.2. PLA nanofibers

3. Biocompatibility of PHA Nanofibers
3.1. Enzymatic degradation of P(3HB) nanofibers
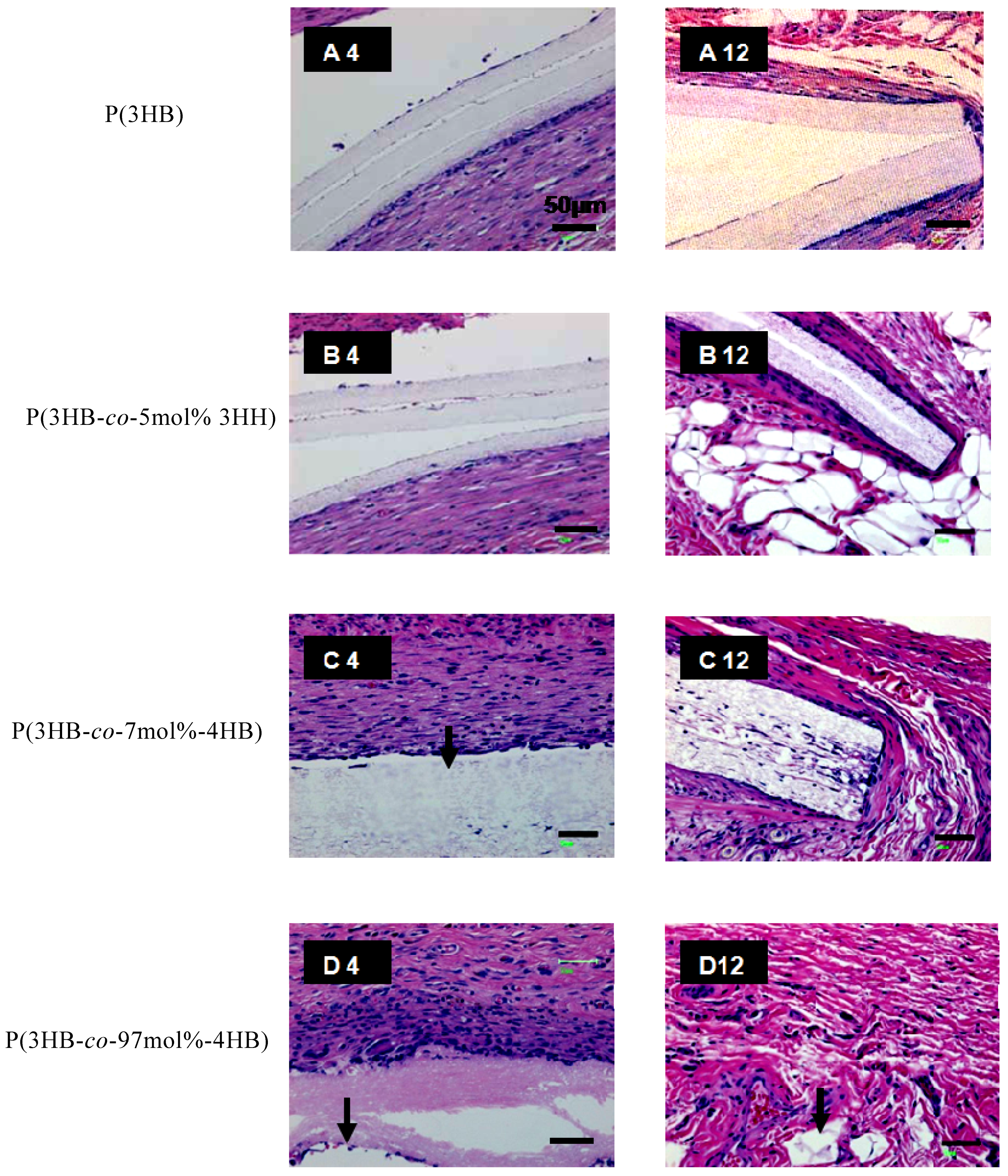
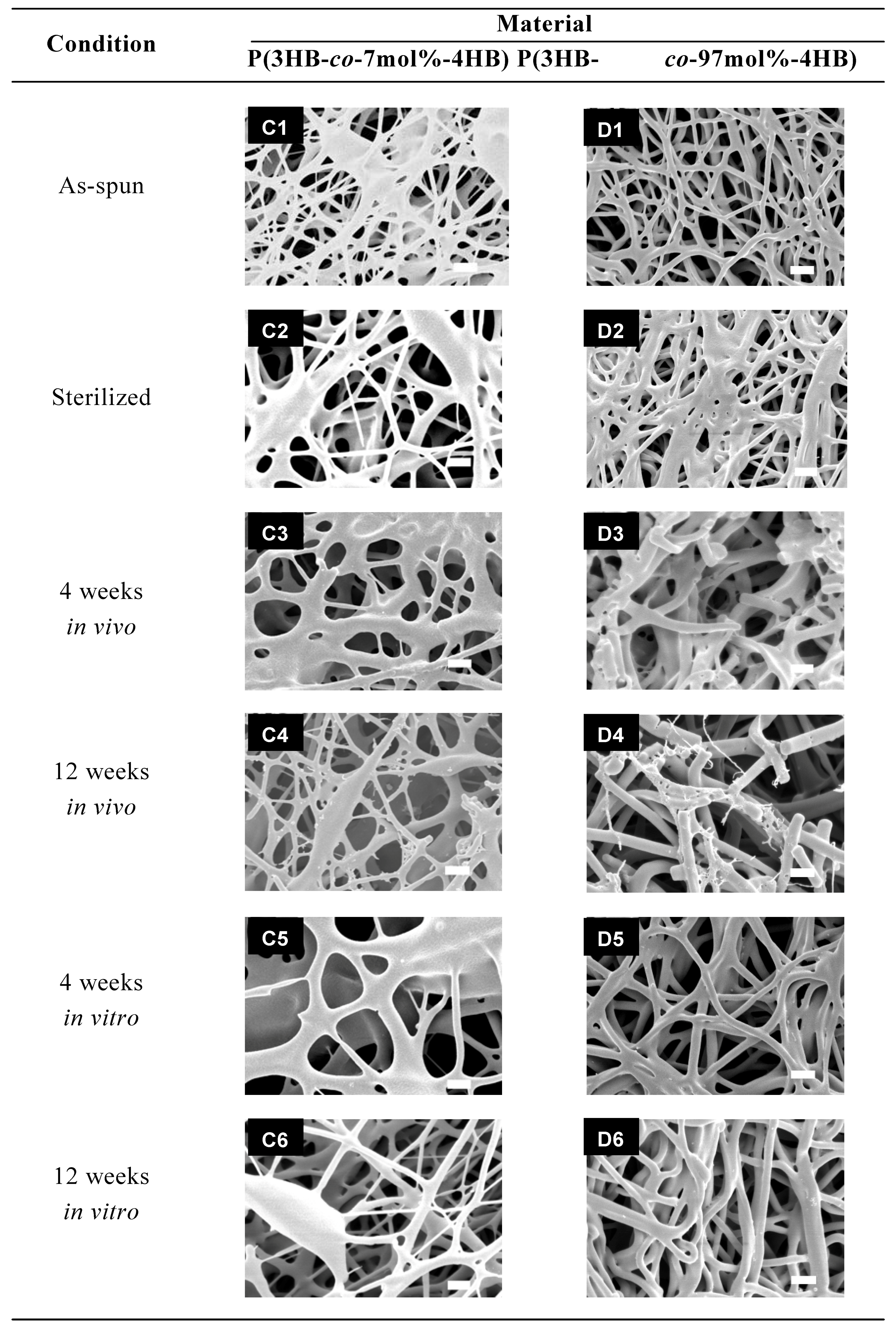
3.2. In vivo and in vitro degradation of PHA nanofibers





| Material | Condition | Mn x 105 | Mw / Mn |
|---|---|---|---|
| P(3HB) | As-spun | 3.5 | 3.1 |
| Sterilized | 2.8 | 3.3 | |
| 4 weeks in vivo | 4.6 | 2.6 | |
| in vitro | 4.2 | 2.6 | |
| 12 weeks in vivo | 2.9 | 2.9 | |
| in vitro | 5.2 | 3.3 | |
| P(3HB-co-5mol%-3HH) | As-spun | 3.6 | 3.6 |
| Sterilized | 3.3 | 3.6 | |
| 4 weeks in vivo | 3.9 | 3.1 | |
| in vitro | 2.6 | 4.3 | |
| 12 weeks in vivo | 3.3 | 3.3 | |
| in vitro | 3.0 | 4.3 | |
| P(3HB-co-7mol%-4HB) | As-spun | 2.3 | 3.0 |
| Sterilized | 2.3 | 3.0 | |
| 4 weeks in vivo | 2.5 | 2.6 | |
| in vitro | 2.3 | 2.6 | |
| 12 weeks in vivo | 1.4 | 2.7 | |
| in vitro | 1.9 | 2.6 | |
| P(3HB-co-97mol%-4HB) | As-spun | 1.1 | 1.5 |
| Sterilized | 1.1 | 1.8 | |
| 4 weeks in vivo | 0.6 | 1.6 | |
| in vitro | 1.3 | 2.0 | |
| 12 weeks in vivo | 0.6 | 2.0 | |
| in vitro | 1.2 | 1.8 |

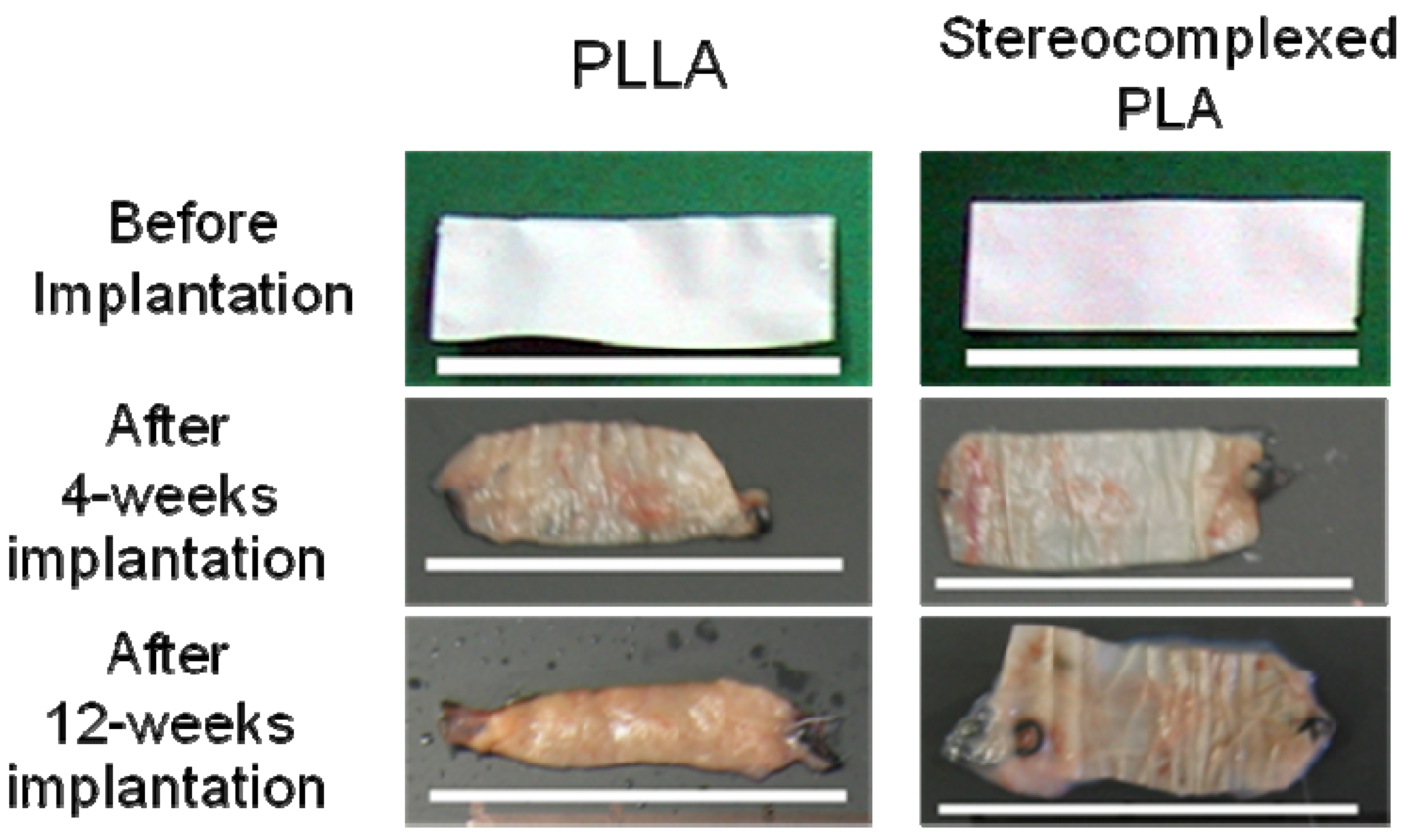
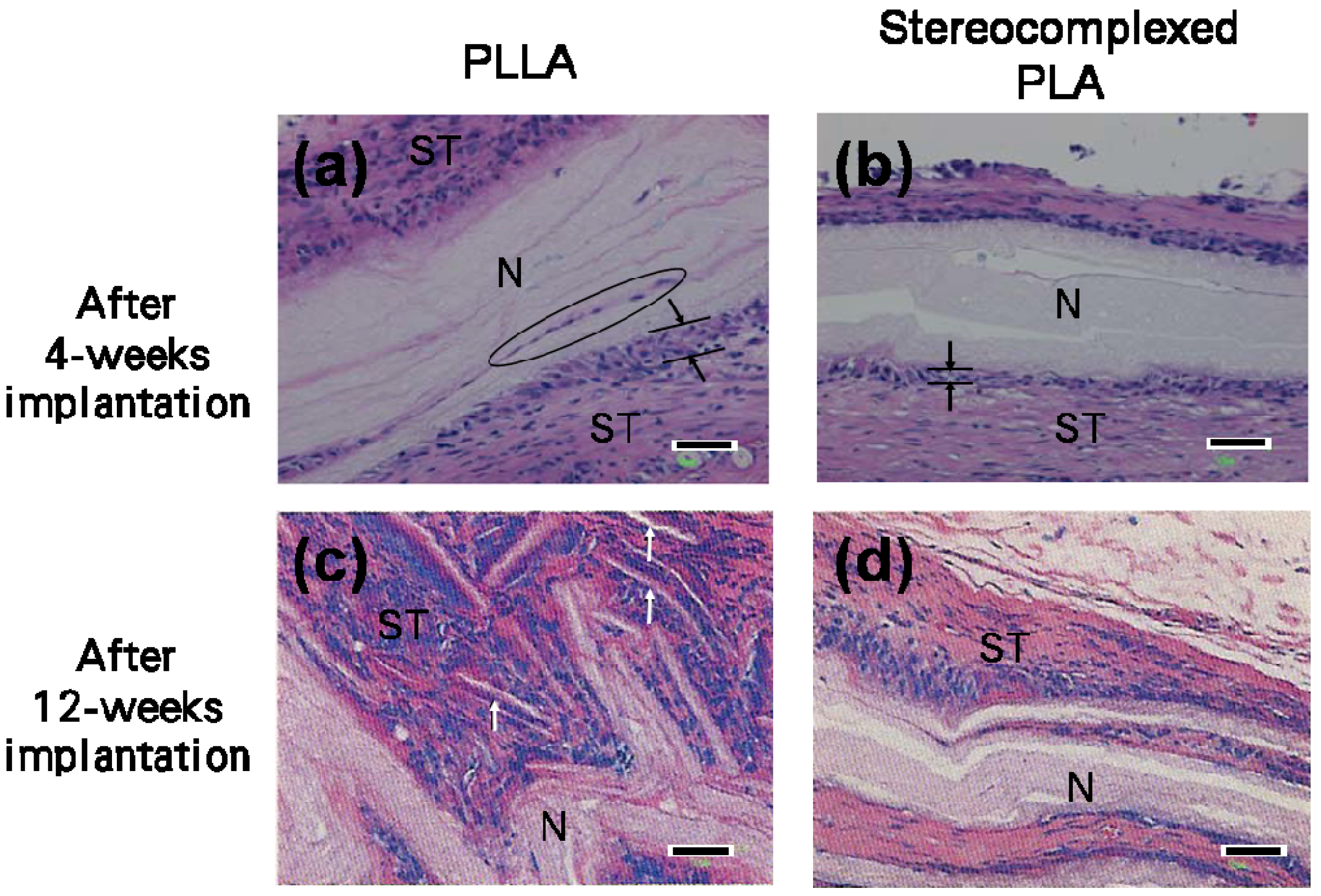
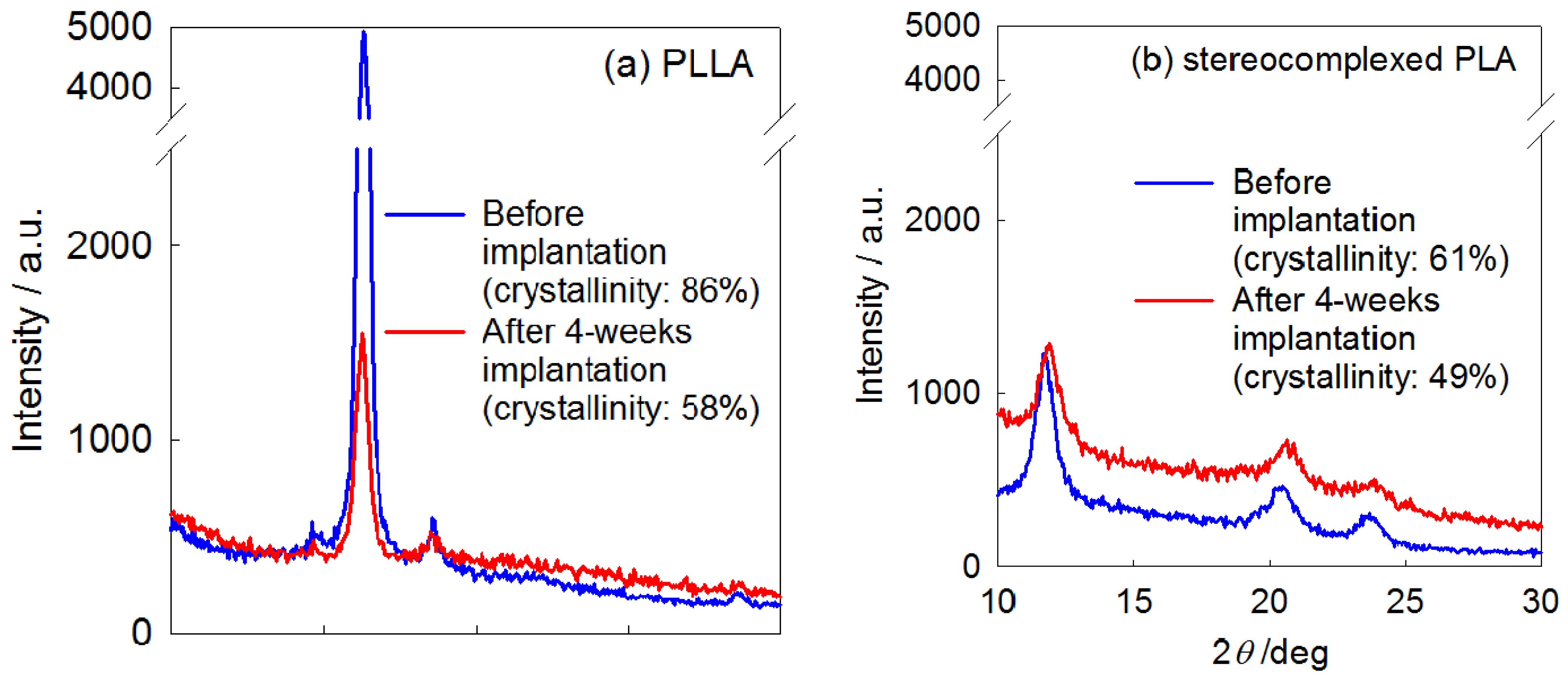
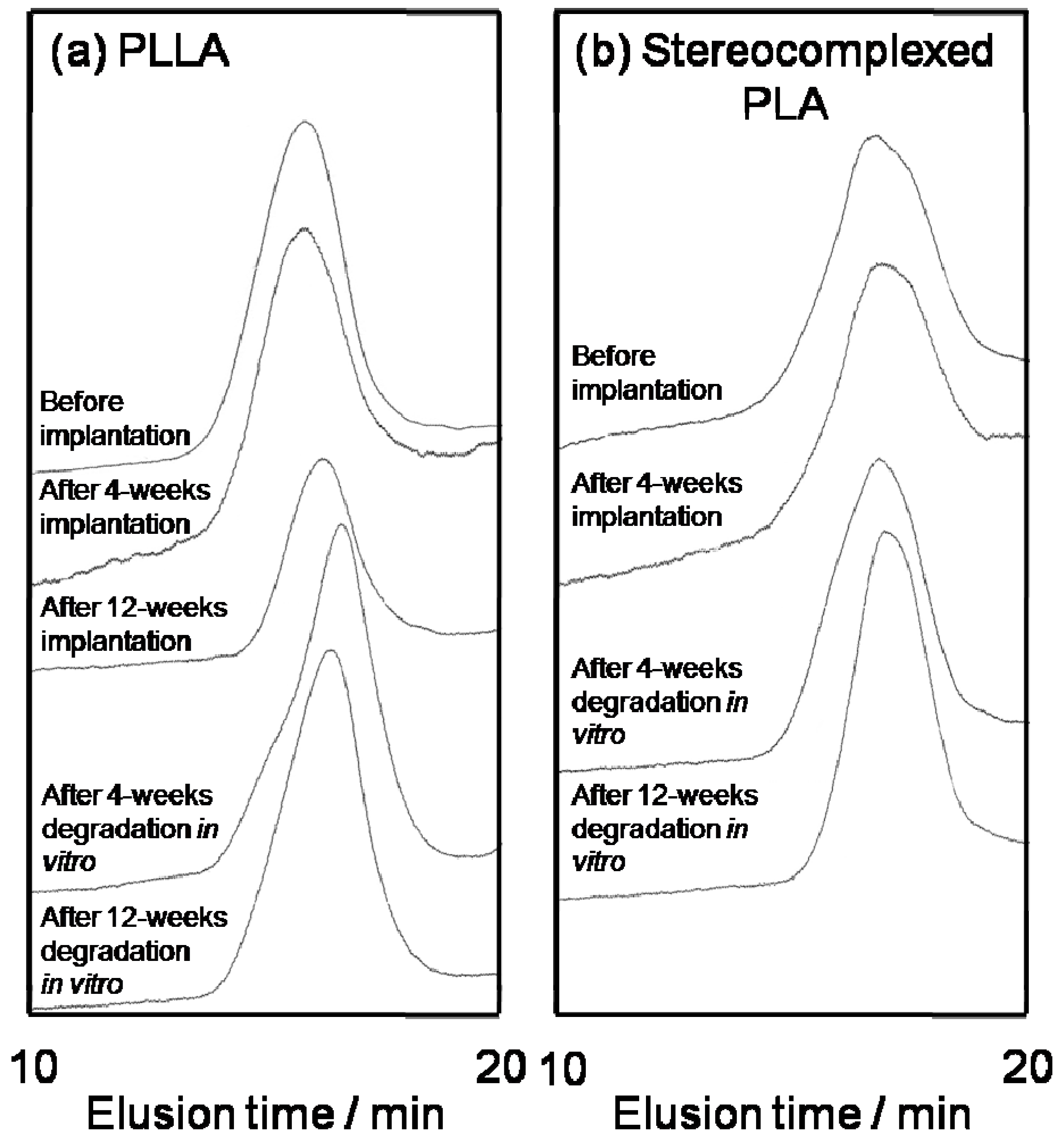
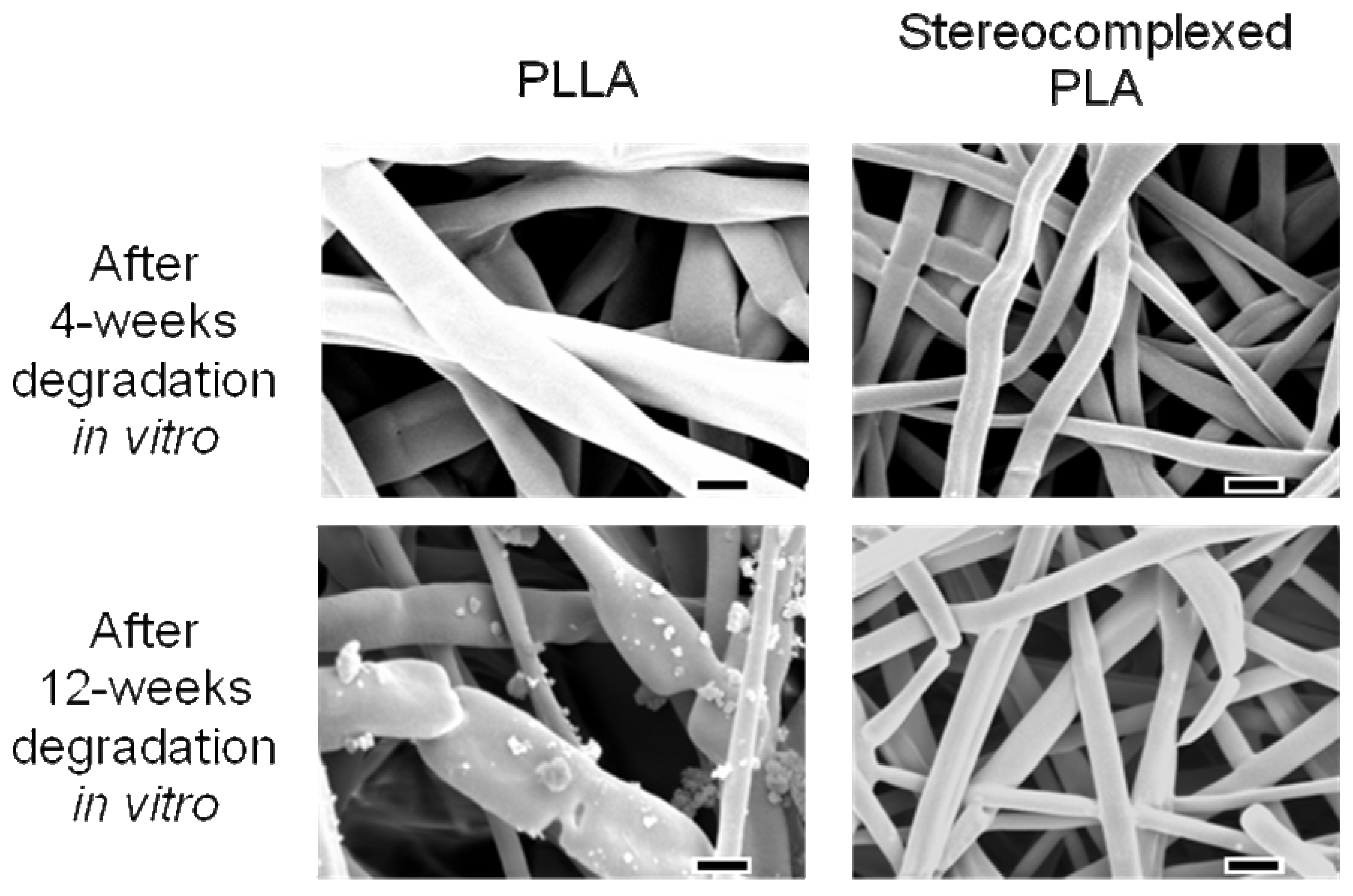
4. Biocompatibility of PLA Nanofibers




| PLLA | PDLA | Stereocomplexed PLA | ||||
|---|---|---|---|---|---|---|
| Mn | Mw/Mn | Mn | Mw/Mn | Mn | Mw/Mn | |
| Original | 4.7 × 105 | 1.8 | 2.2 × 105 | 1.5 | — | — |
| Nanofiber Before implantation | 3.8 × 105 | 2.3 | — | — | 8.7 × 104 | 3.3 |
| After 4-weeks implantation | 3.0 × 105 | 2.4 | — | — | 8.6 × 104 | 2.3 |
| After 12-weeks implantation | 1.7 × 105 | 2.3 | — | — | —a | —a |

| PLLA | Stereocomplexed PLA | |||
|---|---|---|---|---|
| Mn | Mw/Mn | Mn | Mw/Mn | |
| Nanofiber before implantation | 3.8 × 105 | 2.3 | 8.7 × 104 | 3.3 |
| After 4-weeks implantation | 1.4 × 105 | 3.5 | 8.4 × 104 | 3.0 |
| After 12-weeks implantation | 1.8 × 105 | 3.0 | 6.9 × 104 | 2.2 |
5. Conclusions
Acknowledgements
References and Notes
- Langer, R.; Vacanti, J.P. Tissue Engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M. Cell Adhesion: The Molecular Basis of Tissue Architecture and Morphogenesis. Cell 1996, 84, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Tavakoli, T.; Derby, E.; Serebryakova, Y.; Rao, M.S.; Mattson, M.P. Cell-Extracellular Matrix Interactions Regulate Neural Differentiation of Human Embryonic Stem Cells. BMC Dev. Biol. 2008, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Griffith, L.G. Emerging Design Principles in Biomaterials and Scaffolds for Tissue Engineering. Ann N. Y. Acad. Sci. 2002, 961, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Hasirci, V.; Lewandrowski, K.; Gresser, J.D.; Wise, D.L.; Trantolo, D.J. Versatility of Biodegradable Biopolymers: Degradability and an In Vivo Application. J. Biotechnol. 2001, 86, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y. Fermentation and Analysis of Microbial Polyesters. In Microbial Polyesters; VCH Publishers: New York, NY, USA, 1990; pp. 11–31. [Google Scholar]
- Lemoigne, M. Produits de déshydration et de polymérization de l’acide β=oxybutyrique. Bull. Soc. Chim. Biol. 1926, 8, 770–782. [Google Scholar]
- Lenz, R.W.; Marchessault, R.H. Bacterial Polyesters: Biosynthesis, Biodegradable Plastics and Biotechnology. Biomacromolecules 2005, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T. Strong Fibers and Films of Microbial Polyesters. Macromol. Biosci. 2005, 5, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Williams, S.F. Medical Applications of Poly-4-hydroxybutyrate: A Strong Flexible Absorbable Biomaterial. Biochem. Eng. J. 2003, 3738, 1–9. [Google Scholar]
- Hocking, P.J.; Marchessault, R.H. Biopolyesters. In Chemistry and Technology of Biodegradable Polymers; Griffin, G.J.L., Ed.; Chapman & Hall: London, UK, 1994; pp. 48–96. [Google Scholar]
- Williams, S.F.; Martin, D.P. Applications of PHAs in Medicine and Pharmacy. In Biopolymers, 1st ed.; Doi, Y., Steinbüchel, A., Eds.; Wiley-VCH: Weinheim, Germany, 2002; Volume 4, pp. 91–128. [Google Scholar]
- Ikada, Y.; Tsuji, H. Biodegradable Polyesters for Medical and Ecological Applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Iwata, T.; Doi, Y. Alkaline Hydrolysis of Solution-Grown Poly(L-lactic acid) Single Crystals. Sen’i Gakkaishi 2001, 57, 172–177. [Google Scholar] [CrossRef]
- Ikada, Y.; Jamshidi, K.; Tsuji, H.; Hyon, S.H. Stereocomplex Formation between Enantiomeric Poly(lactides). Macromolecules 1987, 20, 904–906. [Google Scholar] [CrossRef]
- Tsuji, H. Poly(lactide) Stereocomplexes: Formation, Structure, Properties, Degradation, and Applications. Macromol. Biosci. 2005, 5, 569–597. [Google Scholar] [CrossRef] [PubMed]
- Okihara, T.; Tsuji, M.; Kawaguchi, A.; Katayama, K.; Tsuji, H.; Hyon, S.H.; Ikada, Y. Crystal Structure of Stereocomplex of Poly(l-lactide) and Poly(d-lactide). J. Macromol. Sci. -Phys. 1991, B30, 119–140. [Google Scholar]
- Tsuji, H. In Vitro Hydrolysis of Blends from Enantiomeric Poly(lactide)s Part 1. Well-Stereo-Complexed Blend and Non-Blended Films. Polymer 2000, 41, 3621–3630. [Google Scholar] [CrossRef]
- Tsuji, H.; Suzuki, M. In Vitro Hydrolysis of Enantiomeric Poly(lactide)s. 2. Well-Stereocomplexed Fiber and Film. Sen’I Gakkaishi 2001, 57, 198–202. [Google Scholar] [CrossRef]
- Tsuji, H.; Miyaushi, S. Enzymatic Hydrolysis of Poly(lactide)s: Effects of Molecular Weight, L-Lactide Content, and Enantiomeric and Diastereoisomeric Polymer Blending. Biomacromolecules 2001, 2, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Reneker, D.H.; Chun, I. Nanometre Diameter Fibres of Polymer, Produced by Electrospinning. Nanotechnology 1996, 7, 216–223. [Google Scholar] [CrossRef]
- Morota, K.; Matsumoto, K.; Mizukoshi, T.; Konosu, Y.; Minagawa, M.; Tanioka, A.; Yamagata, Y.; Inoue, K. Poly(ethylene oxide) Thin Films Produced by Electrospray Deposition: Morphology Control and Additive Effects of Alcohols on Nanostructure. J. Colloid Interface Sci. 2004, 279, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Murugan, R.; Ramakrishna, S. Nano-Featured Scaffolds for Tissue Engineering: A Review of Spinning Methodologies. Tissue Eng. 2006, 12, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Reneker, D.H.; Yarin, A.L.; Fong, H.; Koombhongse, S. Bending Instability of Electrically Charged Liquid Jets of Polymer Solutions in Electrospinning. J. Appl. Phys. 2000, 87, 4531–4547. [Google Scholar] [CrossRef]
- Buchko, C.J.; Chen, L.C.; Shen, Y.; Martin, D.C. Processing and Microstructural Characterization of Porous Biocompatible Protein Polymer Thin Films. Polymer 1999, 40, 7397–7407. [Google Scholar] [CrossRef]
- Zong, X.; Kim, K.; Fang, D.; Ran, S.; Hsiao, B.S.; Chu, B. Structure and Process Relationship of Electrospun Bioabsorbable Nanofiber Membranes. Polymer 2002, 43, 4403–4412. [Google Scholar] [CrossRef]
- Zong, X.; Ran, S.; Kim, K.-S.; Fang, D.; Hsiao, B.; Chu, B. Structure and Morphology Changes during in Vitro Degradation of Electrospun Poly(glycolide-co-lactide) Nanofiber Membrane. Biomacromolecules 2003, 4, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Lee, S.W.; Jeong, L.; Bae, S.-H.; Min, B.C.; Youk, J.H.; Park, W.H. Effect of Organosoluble Salts on the Nanofibrous Structure of Electrospun Poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Int. J. Biol. Macromol. 2004, 34, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Hasuda, H.; Kamitakahara, M.; Ohtsuki, C.; Tanihara, M.; Kang, I.-K.; Kwon, O.H. A Composite of Hydroxyapatite with Electrospun Biodegradable Nanofibers as a Tissue Engineering Material. J. Biosci. Bioeng. 2005, 100, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Sombatmankhong, K.; Suwantong, O.; Waleetorncheepsawat, S.; Supaphol, P. Electrospun Fiber Mats of Poly(3-hydroxybutyrate), Poly(3-hydroxybutyrate-co-3-hydroxyvalerate), and Their Blends. J. Polym. Sci., Part B: Polym. Phys. 2006, 44, 2923–2933. [Google Scholar] [CrossRef]
- Kim, K.; Yu, M.; Zong, X.; Chiu, J.; Fang, D.; Seo, Y.-S.; Hsiao, B.S.; Chu, B.; Hadjiargyrou, M. Control of Degradation Rate and Hydrophilicity in Electrospun Non-Woven Poly(d,l-lactide) Nanofiber Scaffolds for Biomedical Applications. Biomaterials 2003, 24, 4977–4985. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Chen, X.; Liang, Q.; Xu, X.; Jing, X. Enzymatic Degradation of Poly(L-lactide) and Poly(ε-caprolactone) Electrospun Fibers. Macromol. Biosci. 2004, 4, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Min, B.-M.; Lee, S.J.; Lee, T.S.; Park, W.H. In Vitro Degradation Behavior of Electrospun Polyglycolide, Polylactide, and Poly(lactide-co-glycolide). J. Appl. Polym. Sci. 2005, 95, 193–200. [Google Scholar] [CrossRef]
- Orts, W.J.; Marchessault, R.H.; Bluhm, T.L.; Hamer, G.K. Observation of Strain-Induced β-Form in Poly(β-hydroxyalkanoates). Macromolecules 1990, 23, 5368–5370. [Google Scholar] [CrossRef]
- Aoyagi, Y.; Doi, Y.; Iwata, T. Mechanical Properties and Highly Ordered Structure of Ultra-High-Molecular-Weight Poly[(R)-3-hydroxybutyrate] Films: Effects of Annealing and Two-Step Drawing. Polym. Degrad. Stab. 2003, 79, 209–216. [Google Scholar] [CrossRef]
- Iwata, T.; Tsunoda, K.; Aoyagi, Y.; Kusaka, S.; Yonezawa, N.; Doi, Y. Mechanical Properties of Uniaxially Cold-Drawn Films of Poly[(R)-3-hydroxybutyrate]. Polym. Degrad. Stab. 2003, 79, 217–224. [Google Scholar] [CrossRef]
- Iwata, T.; Aoyagi, Y.; Fujita, M.; Yamane, H.; Doi, Y.; Suzuki, Y.; Takeuchi, A.; Uesugi, K. Processing of a Strong Biodegradable Poly[(R)-3-hydroxybutyrate] Fiber and a New Fiber Structure Revealed by Micro-Beam X-Ray Diffraction with Synchrotron Radiation. Macromol. Rapid Commun. 2004, 25, 1100–1104. [Google Scholar] [CrossRef]
- Tanaka, T.; Fujita, M.; Takeuchi, A.; Suzuki, Y.; Uesugi, K.; Ito, K.; Fujisawa, T.; Doi, Y.; Iwata, T. Formation of Highly Ordered Structure in Poly[(R)-3-hydroxybutyrate-co-(R)-3-hydroxyvalerate] Highstrength Fibers. Macromolecules 2006, 39, 2940–2946. [Google Scholar] [CrossRef]
- Iwata, T.; Fujita, M.; Aoyagi, Y.; Doi, Y.; Fujisawa, T. Time-Resolved X-Ray Diffraction Study on Poly[(R)-3-hydroxybutyrate] Films during Two-Step-Drawing: Generation Mechanism of Planar Zigzag Structure. Biomacromolecules 2005, 6, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Nyame, V.; Macdiarmid, A.G., Jr.; Jones, W.E. Polyaniline/Poly(methyl methacrylate) Coaxial Fibers: The Fabrication and Effects of the Solution Properties on the Morphology of Electrospun Core Fibers. J. Polym. Sci., Part B: Polym. Phys. 2004, 42, 3934–3942. [Google Scholar] [CrossRef]
- Lyons, J.; Li, C.; Ko, F. Melt-electrospinning Part I: Processing Parameters and Geometric Properties. Polymer 2004, 45, 7597–7603. [Google Scholar] [CrossRef]
- Fong, H.; Chun, I.; Reneker, D.H. Beaded Nanofibers Formed during Electrospinning. Polymer 1999, 40, 4585–4592. [Google Scholar] [CrossRef]
- Zuo, W.; Zhu, M.; Yang, W.; Yu, H.; Chen, Y.; Zhang, Y. Experimental Study on Relationship between Jet Stability and Formation of Beaded Fibers during Electrospinning. Polym. Eng. Sci. 2005, 45, 704–709. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y.; Hyon, S.H.; Kimura, Y.; Kitao, T. Stereocomplex Formation between Enantiomeric Poly(lactic acid)s. VIII. Complex Fibers Spun from Mixed Solution of Poly(D-lactic acid) and poly(L-lactic acid). J. Appl. Polym. Sci. 1994, 51, 337–344. [Google Scholar] [CrossRef]
- Takasaki, M.; Ito, H.; Kikutani, T. Structure Development of Polylactides with Various D-Lactide Contents in the High-Speed Melt Spinning Process. J. Macromol. Sci., Part B: Phys. 2003, 42, 57–73. [Google Scholar] [CrossRef]
- Furuhashi, Y.; Kimura, Y.; Yamane, H. Higher Order Structural Analysis of Stereocomplex-Type Poly(lactic acid) Melt-Spun Fibers. J. Polym. Sci., Part B: Polym. Phys. 2007, 45, 218–228. [Google Scholar] [CrossRef]
- Tsuji, H.; Nakano, M.; Hashimoto, M.; Takashima, K.; Katsura, S.; Mizuno, A. Electrospinning of Poly(lactic acid) Stereocomplex Nanofibers. Biomacromolecules 2006, 7, 3316–3320. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Aoyagi, Y.; Tanaka, T.; Fujita, M.; Takeuchi, A.; Suzuki, Y.; Uesugi, K. Micro-Beam X-Ray Diffraction and Enzymatic Degradation of Poly[(R)-3-hydroxybutyrate] Fibers with Two Kinds of Molecular Conformations. Macromolecules 2006, 39, 5789–5795. [Google Scholar] [CrossRef]
- Rappolee, D.A.; Mark, D.; Banda, M.J.; Werb, Z. Wound Macrophages Express TGF-alpha and Other Growth Factors in Vivo: Analysis by mRNA Phenotyping. Science 1988, 241, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Skraly, F.A.; Williams, S.F. Polyhydroxyalkanoate Compositions Having Controlled Degradation Rats. PCT Patent Application No. WO 99/32536, 1999. [Google Scholar]
- Qu, X.H.; Wu, Q.; Zhang, K.Y.; Chen, G.Q. In Vivo Studies of Poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) Based Polymers: Biodegradation and Tissue Reactions. Biomaterials 2006, 27, 3540–3548. [Google Scholar] [PubMed]
- Ishii, D.; Lee, W.-K.; Kasuya, K.; Iwata, T. Fine Structure and Enzymatic Degradation of Poly[(R)-3-hydroxybutyrate] and Stereocomplexed Poly(lactide) Nanofibers. J. Biotechnol. 2007, 132, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Robertson, J.L.; Spillman, W.B., Jr.; Claus, R.O. Effects of the Chemical Structure and the Surface Properties of Polymeric Biomaterials on Their Biocompatibility. Pharm. Res. 2004, 21, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Ishii, D.; Mahara, A.; Murakami, S.; Yamaoka, T.; Sudesh, K.; Samian, R.; Fujita, M.; Maeda, M.; Iwata, T. Scaffolds from Electrospun Polyhydroxyalkanoate Copolymers: Fabrication, Characterization, Bioabsorption and Tissue Response. Biomaterials 2008, 29, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Ishii, D.; Tang, H. Y.; Mahara, A.; Murakami, S.; Yamaoka, T.; Iwata, T. In Vivo Tissue Response and Degradation Behavior of PLLA and Stereocomplexed PLA Nanofibers. Biomacromolecules 2009, 10, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Bien, H.; Chung, C.-Y.; Yin, L.; Fang, D.; Hsiao, B.S.; Chu, B.; Entcheva, E. Electrospun Fine-Textured Scaffolds for heart Tissue Constructs. Biomaterials 2005, 26, 5330–5338. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishii, D.; Ying, T.H.; Yamaoka, T.; Iwata, T. Characterization and Biocompatibility of Biopolyester Nanofibers. Materials 2009, 2, 1520-1546. https://doi.org/10.3390/ma2041520
Ishii D, Ying TH, Yamaoka T, Iwata T. Characterization and Biocompatibility of Biopolyester Nanofibers. Materials. 2009; 2(4):1520-1546. https://doi.org/10.3390/ma2041520
Chicago/Turabian StyleIshii, Daisuke, Tang Hui Ying, Tetsuji Yamaoka, and Tadahisa Iwata. 2009. "Characterization and Biocompatibility of Biopolyester Nanofibers" Materials 2, no. 4: 1520-1546. https://doi.org/10.3390/ma2041520
APA StyleIshii, D., Ying, T. H., Yamaoka, T., & Iwata, T. (2009). Characterization and Biocompatibility of Biopolyester Nanofibers. Materials, 2(4), 1520-1546. https://doi.org/10.3390/ma2041520