1. Introduction
There are conflicting reports in the literature addressing the question whether the amino acid (AA) sequence of proteins that fold into well-defined structure is recognizably different from those that are randomly generated, as reviewed by De Lucrezia et al. [
1]. However, recent work [
2] has shown that there is a significant difference in the relative propensities of AA triplets and quadruplets in sequences that represent actually folded proteins with a known structure and sequences that were generated randomly, even if the randomly generated sequences followed the natural amino acid propensities.
By distilling the propensity statistics into a score for a given sequence this relative propensity difference was then used to make rather reliable predictions about the foldability of sequences not used in the development of the statistics. However, no significant correlation was found between the stability of a protein and its score.
The previous work was limited to triplets and quadruplets since even for quadruplets some of the 204 sequences were absent in the sequence set of proteins with known structure that was used in the development of the score. The present work is based on recognizing the fact that the absence of an n-tuple in the sequences of folded proteins is also significant information thus it is worth examining what can be learned from the, obviously limited, statistics of AA pentuplets, hextuplets and heptuplets.
Accurate prediction of the change in stability of a protein as a result of mutation is of fundamental interest both for the understanding of the mechanisms of disease-causing mutation and for the field of protein engineering. Given the complexity of proteins and their environment, quantitative predictions require statistical-mechanical approaches, namely, free-energy simulations generally requiring long calculations on hundreds of processors (for a review, see Reference [
3]). Thus computationally simple approaches, even if of lower accuracy, can be of significant interest.
The previous work also compared properties of folded proteins and intrinsically disordered proteins (IDP). As only small differences were observed, given the low precision of the data that the present paper is dealing with, IDPs are not considered in this work.
Furthermore, there is a vast literature on the structural implication of various sequence motifs, including various attempts to predict structure from sequence. Given that the number of relevant protein sequences significantly outnumbers the number of proteins with a known structure, there is merit in studying the properties of protein sequences without including structural information, the aim of the present work.
It will be shown that the pentuplets statistics, despite not sampling over 30% of the pentuplet space, is able to provide an even better separation of folding sequences from the randomly generated ones than the quadruplet statistics-based score used in Reference [
2]. Furthermore, it will be shown that a simplified score, based on the limited hextuplet statistics and on the very limited heptuplet statistics is capable to produce useful prediction for the direction of stability change of a protein upon a mutation.
2. Materials and Methods
As described in Reference [
2], the sequences of the structures in the Protein Data bank (PDB) [
4] in 2018 were downloaded as the file ss.txt from the PDB website,
https://www.rcsb.org/. Filtering out sequences over 50% sequence identity and fewer than 20 residues resulted in 35,667 sequences, referred to here as the experimental or PDB set. Furthermore, histidine tags were removed before each analysis. The set of sequences of a separate set of structures, deposited after the ss.txt file was also generated from the PDB to be used for the prediction of foldability; it will be referred to as the new PDB set.
Two randomly generated sets were used. As described in Reference [
2], one set sampled the amino acids from the uniform distribution (referred to as RU set) and the other with the probability of their respective natural propensities (referred to as RW set).
The full sequences of proteins in the dataset of mutations [
5] were matched to the sequences in the file ss.txt. Finding the correct match required adding the specification of starting residue IDs in the file ss.txt to the dataset—this was done manually.
The score developed in Reference [
2] for triplets and quadruplets has been applied to pentuplets in the present work, defined as follows. For each sequence and each construct
p (triplet, quadruplet and pentuplet) the logarithmic score
SCp was defined as:
where
N is the number of constructs in the sequence and
PNi and
PRi are the probabilities of finding the construct
i in the experimental set and in the
RW set, resp. For constructs
i that were missing from the experimental set
PNi was set to 0.5/20
f (
f = 3, 4 and 5 for triplets, quadruplets and pentuplets, resp.).
For hexamers and heptamers, where the sequences actually seen in the set of folded proteins used is a small fraction of the total number, a simpler score
SSCp was defined:
where
ni is the number of occurrences of the construct
i in the experimental set. This choice was motivated by the fact that the hextuplet and heptuplet counts are so sparse that they can not be considered to be a reasonable approximation of their probability of occurrence.
It should be noted that the problem of sparse statistics can be also dealt with using a reduced AA alphabet, as done, e.g., in [
6]. For the study of mutations, however, it would be of limited use since a significant number of mutations would not represent change in terms of the reduced representation.
While the simple score
SSCp was used only to study mutations, the pentuplet score
SCp was also tested for its ability to predict the foldability of a sequence employing the methods used in Reference [
2]. This test consisted of the following steps:
Determine the distribution of scores over the PDB set.
Determine the distribution of scores over the RW set, consisting of 100,000 sequences of 200 residues.
Calculate the overlap between the two distributions to see how well the scores can distinguish between folding and non-folding sequences.
For a given sequence, calculate its score and see where it lies with respect to the score value at the intersection of the two distributions, resulting in the prediction regarding the foldability of that sequence.
For the study of a mutation using the n-tuplet statistics (n = 3, 4, 5, 6, 7), the scores of all n-tuplets that included the mutated residue were averaged before and after the mutation. The change in the scores was then compared with the change in melting temperatures that are diagnostic of the stability of the protein in question. The comparison included calculating the Pearson correlation between the score change and the melting temperature change and the match in sign change between the score change and the melting temperature change.
The calculations were performed by the program Fold, available at the URL
http://inka.mssm.edu/~mezei/fold, free to academic use; commercial users are charged a nominal fee. The amino acid propensities used are incorporated into the program. The PDB sequence, the new PDB sequence set, and the mutation data set from Reference [
5], extended with the initial residue ID of the protein in the PDB [
4] are also available there.
3. Results
In addition to the existing statistics on triplets and quadruplets, the same data was collected for pentamers, hexamers and heptamers on the experimental set. The coverage of the multiplet space was 100% for triplets, 99.58% for quadruplets, 69.92% for pentuplets, 11.33% for hextuplets and 0.68% for heptuplets. The new statistics were then used for (a) the prediction of foldability of a sequence and (b) prediction of the direction the stability of a protein would change upon mutation.
3.1. Foldability Prediction Using Pentuplet Statistics
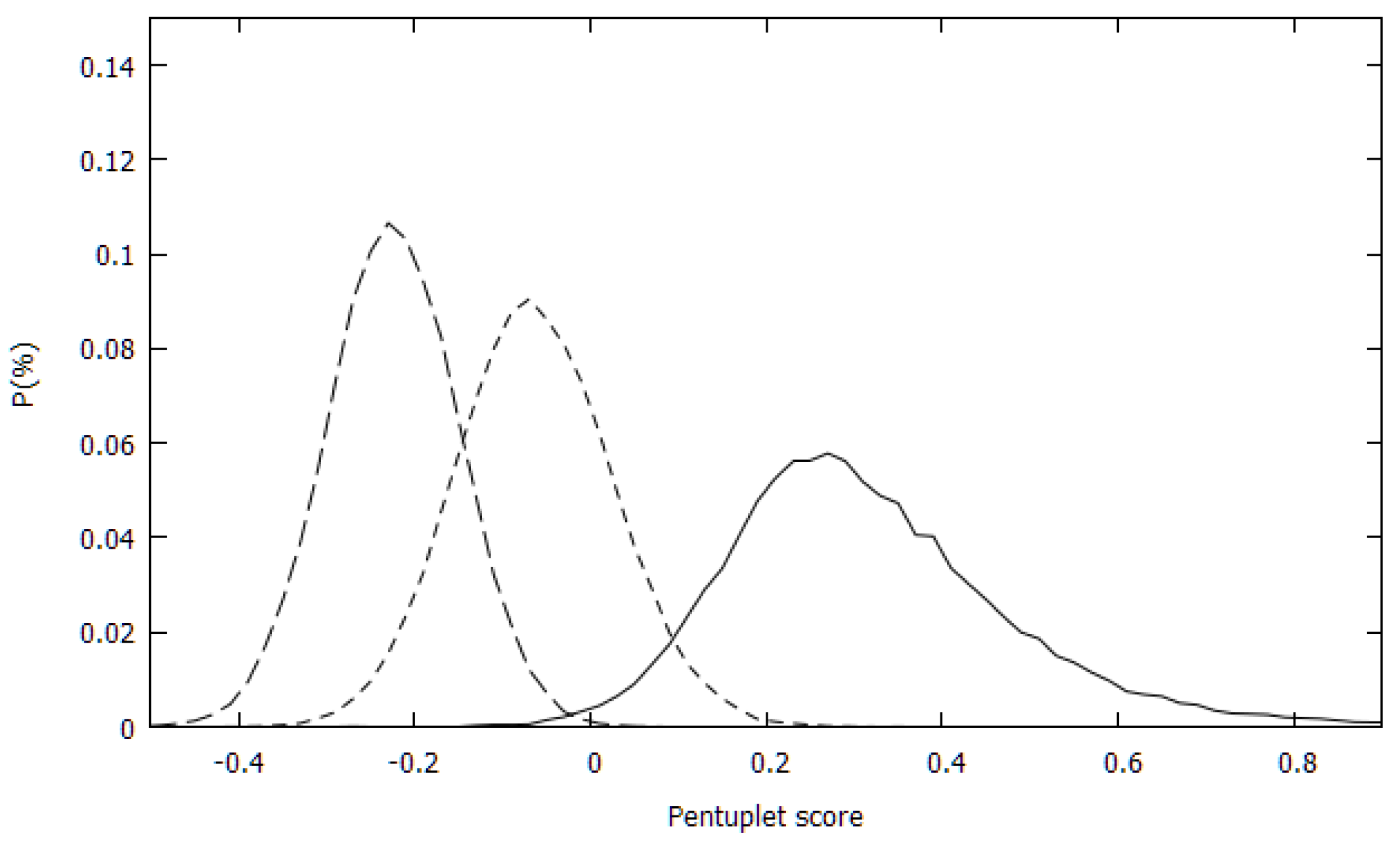
The distributions of the pentuplet scores for the PDB set and for the two randomly generated sets (RW and RU) were first calculated.
Figure 1 shows the three distributions. There was an almost complete separation between the experimental and RW sets’ distributions and a rather small overlap between the distributions of the experimental and RU sets.
To quantitate this observation, the overlaps between the distributions were also calculated. They are 0.096, 0.009 and 0.324 for the PDB-RU, PDB-RU and RU-RW distribution pairs. For comparison, the overlaps between the quadruplets scores of the PDB and the random sets were 0.142 and 0.191 for the PDB-RU and PDB-RW, resp. Unlike the distributions of the quadruplet scores where the RU and RW distributions were nearly identical with 0.786 overlap, the two pentuplet-based random distributions have rather small overlaps. For triplets and quadruplets the PDB-RW overlap was also larger than the PDB-RU overlap, conforming to the expectation that the RW set should be more like the PDB set than the RU set. For the pentuplet score distribution, however, this relation was reversed. This was likely an artifact of the treatment of the missing statistics in the present approach.
To test the predictive power of the pentuplet-based score, the scores of the new PDB set (that were not used for the development of the scores) were compared to newly generated RU and RW sets. A score was considered diagnostic of foldability if it was larger than the score where the PDB and random score distributions intersected, otherwise it was considered diagnostic of randomness.
Table 1 summarizes the predictions using the quadruplet-based predictions from Reference [
2] (using the RW set as reference) and the new pentuplet-based predictions using the both the RU set and the RW set as reference.
The comparison is mixed between the quadruplet and pentuplet scores. For randomly generated sets the pentuplet scores were significantly better than the quadruplet scores. However, the pentuplet scores missed predicting foldable significantly more of the PDB set than the quadruplet score when the RU-based pentuplet distribution was used; using the RW-based distribution the number of missed predictions in the PDB set was essentially the same for the quadruplet and pentuplet scores.
The fact that predictions on the randomly generated sets gave very few false negatives (i.e., if a sequence was predicted to be foldable it was most likely correct) suggested the combination of the quadruplet scores and pentuplet scores in the following way: whenever the quadruplet score predicts randomness, but the pentuplet score predicts foldability, change the prediction to foldable; otherwise keep the quadruplet score prediction. Given the large difference in the distributions of the two randomly generated sets, combination with both reference pentuplet distributions were tested. The resulting predictions are collected in
Table 2.
Using the pentuplet-adjusted quadruplet scores indeed reduced the chance that a foldable sequence would be predicted as non foldable. On the other hand, the predictions on randomly generated sets became less reliable than the predictions based on pentuplet scores only; while using the RU reference distribution resulted in the same prediction accuracy as using quadruplet scores only; using the RW reference set resulted in significantly worse predictions on the set generated randomly from the uniform AA distribution.
3.2. Prediction of Protein Stability Change Upon Mutationon
The mutations in the dataset [
5] were used to calculate the change in the score averages over
n-tuplet scores of
n-tuplets that included the mutated residue. Altogether 1674 mutations were used. For triplets, quadruplets and pentuplets the logarithmic score SC
p (Equation (1)) was used while for the hextuplet and heptuplet scores SSC
p, the simplified formula of Equation (2), was used.
The Pearson correlation between the
n-tuplet scores (
n = 3, 4, 5, 6, 7) were −0.04, 0.03, 0.11, 0.10 and 0.18, resp. While there was a very weak trend toward stronger correlation with increasing
n, the correlation between the score changes and melting temperature changed followed the pattern seen in Reference [
2] for the correlation between scores of the full sequence and the melting temperature of the protein, i.e., virtually no correlation.
However, when the comparison was restricted to the sign changes of scores and melting temperatures, a useful pattern emerged, as seen in the data of
Table 3. Using triplet or quadruplet scores or their combination found sign matches barely above 50% of the mutations, thus having virtually no predictive power. The results for pentuplets, hextuplets and heptuplets were more promising as they show matches around 70%, with the most matches seen with the hextuplets. Different combinations of pentuplets, hextuplets and heptuplets all showed a few % more matches, although only on a limited number of mutations where there was a consensus of the score sign changes among the
n-tuplets considered.
4. Discussion
Motivated by the success of the sequence-based predictions for the foldability of an AA sequence using statistics of triplets and quadruplets [
2] the present work examined the use of longer
n-tuplets. Since for longer
n-tuplets the statistics gets progressively sparser the treatment of missing or low-quality statistics needed newer approaches.
The quality of the statistics over pentuplets was the closest to the quality of the triplet and quadruplets statistics so an attempt was made to use them for foldability prediction. As a warning sign that the missing information has significant effect, the score distributions of the two kinds of randomly generated sets were very different and, counterintuitively, the distribution of the AA-propensity based set was more different from the PDB set than distribution of the uniform AA distribution based set. While the predictions on random sets improved, the prediction on the new PDB set worsened, especially when the RU reference set was used.
The high quality of the pentuplets predictions on the randomly generated sets suggested a combination of the quadruplet and pentuplets prediction. It was found that if the quadruplet predictions were modified with the pentuplets prediction using the RU reference distribution, the quality of predictions on the random sets remained at the quadruplet level but on the new PDB set it improved significantly. This combination was suggested to be an improvement over the quadruplet-based predictions.
The samples of hextuplets and heptuplets were way too sparse to give estimates of probabilities. Thus a score using only simple counts was developed and used for a different purpose, that of predicting if a mutation increased the stability of a protein or not.
The prediction using pentuplet, hextuplet and heptuplet statistics were all found to predict the stability change direction of ~70% of the mutations examined. Limiting predictions to mutations where more than one n-tuplets gave the same prediction slightly improved the prediction accuracy: the best prediction accuracy (73.4%) was obtained when pentuplets, hextuplets and heptuplets were combined. While this was still rather weak, given the minimal computing expense involved, especially in comparison to the computational cost of free-energy simulations, it could be helpful in screening a large set of putative mutations for experimental tests.
If Reference [
2] the predictions were based on the statistics of two single properties, the distributions of AA triplets and quadruplets. Several other properties showed differences between the PDB set and the randomly generated sets but not used for foldability predictions as the differences were smaller. The present work showed that some combination of scores resulted in improved prediction accuracy. Future work is planned to find ways of incorporating the statistics so far ignored to further enhance the accuracy of foldability prediction.




{kind=link}