
Prediction of Intrinsically Disordered Proteins Using Machine Learning Algorithms Based on Fuzzy Entropy Feature

Abstract
:1. Introduction
2. Features Selection of IDPS
2.1. Shannon Entropy
2.2. Topological Entropy
2.3. Fuzzy Entropy
2.4. Amino Acid Propensity Scale
3. Algorithm Principle
3.1. Linear Discriminant Analysis
3.2. Support Vector Machines
3.3. Back Propagation Neural Network
4. Performance Evaluation
5. Data Preprocessing Method
6. Result and Discussion
6.1. The Simulation Results
6.2. The Influence of Fuzzy Entropy on Prediction Effect
7. Conclusions
- 1.
- In the sequence identification of intrinsically disordered proteins, this paper extracts feature sets based on topological entropy, Shannon entropy, fuzzy entropy and information from two amino acid propensity tables. The multiple perspectives of information extraction can lead to a better representation of the sequence information of proteins. Among them, fuzzy entropy is applied as a feature for the first time in this field.
- 2.
- In response to the uneven distribution of the dataset and the poor homogeneity of the dataset as a whole, we have used a windowing approach to the dataset. After comparison, the windowed data is more robust and has a more concentrated data distribution, and is more accurate for predicting inherently disordered proteins.
- 3.
- After comparison, the performance of all three algorithms we constructed was improved after applying fuzzy entropy as a feature. Moreover, the recognition accuracy of our algorithms is as accurate as several current algorithms. Among the three schemes we constructed, BP-FE performs the best, and the MCC can reach 0.51, which exceeds many existing schemes. It is worth noting that the algorithm that uses fuzzy entropy as a feature requires only five features, whereas most algorithms used as a comparison require more than 30 features to make predictions.
- 1.
- Although this paper has achieved good results for the identification of intrinsically disordered proteins, a classification model with higher prediction accuracy and more practical significance is needed for practical applications. Therefore, in future studies on intrinsically disordered proteins, more in-depth studies will be carried out with the aim of further improving the MCC by considering more influencing factors as a condition. For example, amino acids possess more than 500 physicochemical properties, can these five features we have chosen contain information on all aspects of amino acids? This is a question that we need to focus on in our future work.
- 2.
- As classification of intrinsically disordered proteins is a more fundamental part of proteomics, deeper exploration is essential in order to take full advantage of the unique biological functions of each intrinsically disordered protein. Future research may revolve around exploring the interactions between intrinsically disordered proteins, protein-ligand interactions, etc.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nordberg, R.C.; Loboa, E.G. Our Fat Future: Translating Adipose Stem Cell Therapy. Stem Cells Transl. Med. 2015, 4, 974–979. [Google Scholar] [CrossRef]
- Lieutaud, P.; Ferron, F.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. How disordered is my protein and what is its disorder for? A guide through the “dark side” of the protein universe. Intrinsically Disord. Proteins 2016, 4, e1259708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Ann. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence Complexity of Disordered Protein. Proteins Struct. Funct. Bioinform. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Rune, L.; Russell, R.B.; Victor, N.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar]
- Zsuzsanna, D.; Tompa, P.; Simon, I. Prediction of protein disorder at the domain level. Curr. Protein Peptide Sci. 2007, 8, 161–171. [Google Scholar]
- Jaime, P.; Felder, C.E.; Tzviya, Z.B.M.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndexl©: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar]
- Lobanov, M.Y.; Galzitskaya, O.V. The Ising model for prediction of disordered residues from protein sequence alone. Phys. Biol. 2011, 8, 035004. [Google Scholar] [CrossRef] [PubMed]
- PONDR: Predictors of Natural Disordered Regions. Available online: http://www.pondr.com/ (accessed on 12 June 2007).
- Shimizu, K.; Hirose, S.; Noguchi, T. POODLE-S: Web application for predicting protein disorder by using physicochemical features and reduced amino acid set of a position-specific scoring matrix. Bioinformatics 2007, 23, 2337. [Google Scholar] [CrossRef]
- Hirose, S.; Shimizu, K.; Kanai, S.; Kuroda, Y.; Noguchi, T. POODLE-L: A two-level SVM prediction system for reliably predicting long disordered regions. Bioinformatics 2007, 23, 2046–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Muraoka, Y.; Hirose, S.; Tomii, K.; Noguchi, T. Predicting mostly disordered proteins by using structure unknown protein data. BMC Bioinform. 2007, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.J.; Mcguffin, L.J.; Bryson, K.; Buxton Bernard, F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef]
- Medina, M.W.; Gao, F.; Naidoo, D.; Rudel, L.L.; Temel, R.E.; McDaniel, A.L.; Marshall, S.M.; Krauss, R.M. Coordinately Regulated Alternative Splicing of Genes Involved in Cholesterol Biosynthesis and Uptake. PLoS ONE 2011, 6, e19420. [Google Scholar] [CrossRef] [Green Version]
- Chung-Tsai, S.; Chien-Yu, C.; Chen-Ming, H. iPDA: Integrated protein disorder analyzer. Nucleic Acids Res. 2007, 35, 465–472. [Google Scholar]
- Tompa, M.F.P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef]
- Alessandro, V.; Oscar, B.; Gianluca, P.; Tosatto, S.C.E. Spritz: A server for the prediction of intrinsically disordered regions in protein sequences using kernel machines. Nucleic Acids Res. 2006, 34, 164–168. [Google Scholar]
- Su, C.T.; Chen, C.Y.; Ou, Y.Y. Protein disorder prediction by condensed PSSM considering propensity for order or disorder. BMC Bioinform. 2006, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mizianty, M.J.; Stach, W.; Chen, K.; Kedarisetti, K.D.; Disfani, F.M.; Kurgan, L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, i489–i496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcguffin, L.J. Intrinsic disorder prediction from the analysis of multiple protein fold recognition models. Bioinformatics 2008, 24, 1798–1804. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.R.; Thomson, R.; Mcneil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, I.E.; Ibrikci, T.; Ersoy, O.K. Prediction of disorder with new computational tool: BVDEA. Exp. Syst. Appl. 2011, 38, 14451–14459. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Zhao, J. A Low Computational Complexity Scheme for the Prediction of Intrinsically Disordered Protein Regions. Math. Probl. Eng. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, X.; Liu, B. IDPCRF: Intrinsically Disordered Protein/Region Identification Based on Conditional Random Fields. Int. J. Mol. Sci. 2018, 19, 2483. [Google Scholar] [CrossRef] [Green Version]
- Megan, S.; Hamilton, J.A.; Tanguy, L.G.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The Database of Disordered Proteins. Nucleic Acids Res. 2007, 35, 786–793. [Google Scholar]
- Wei, L.; Ding, Y.; Su, R.; Tang, J.; Zou, Q. Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 2017, 117, 212–217. [Google Scholar] [CrossRef]
- Lee, K.; Do, D.T.; Hung, T.N.K.; Lam, L.H.T.; Huynh, T.; Nguyen, N.T.K. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int. J. Mol. Sci. 2020, 21, 9070. [Google Scholar] [CrossRef]
- Lam, L.H.T.; Le, N.H.; van Tuan, L.; Tran Ban, H.; Nguyen Khanh Hung, T.; Nguyen, N.T.K.; Huu Dang, L.; Le, N.Q.K. Machine Learning Model for Identifying Antioxidant Proteins Using Features Calculated from Primary Sequences. Biology 2020, 9, 325. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | R | N | D | C | Q | E | G | H | K | |
| Mapping values | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| M | P | S | T | I | L | F | W | Y | V | |
| Mapping values | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | R | N | D | C | Q | E | G | H | K | |
| Mapping values | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| M | P | S | T | I | L | F | W | Y | V | |
| Mapping values | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| A | R | N | D | C | Q | E | G | H | I | |
| Remarking465 | −0.0537 | −0.2141 | 0.2911 | −0.5301 | 0.3088 | 0.5214 | 0.0149 | 0.1696 | 0.2907 | 0.1739 |
| Bfactor(2STD) | 0.0633 | 0.2120 | 0.3480 | −0.4940 | 0.1680 | 0.4560 | 0.1060 | −0.0910 | −0.1400 | −0.4940 |
| L | K | M | F | P | S | T | W | Y | V | |
| Remarking465 | −0.3379 | 0.1984 | −0.1113 | −0.8434 | −0.0558 | 0.2627 | −0.1297 | −1.3710 | −0.8040 | −0.2405 |
| Bfactor(2STD) | −0.3890 | 0.4020 | −0.1260 | −0.5260 | 0.1800 | 0.1260 | −0.0390 | −0.7260 | −0.5060 | −0.4630 |
| LDA | SVM | BP | |
|---|---|---|---|
| Before windowed | 0.3769 | 0.4109 | 0.4483 |
| After windowed | 0.4396 | 0.4818 | 0.4953 |
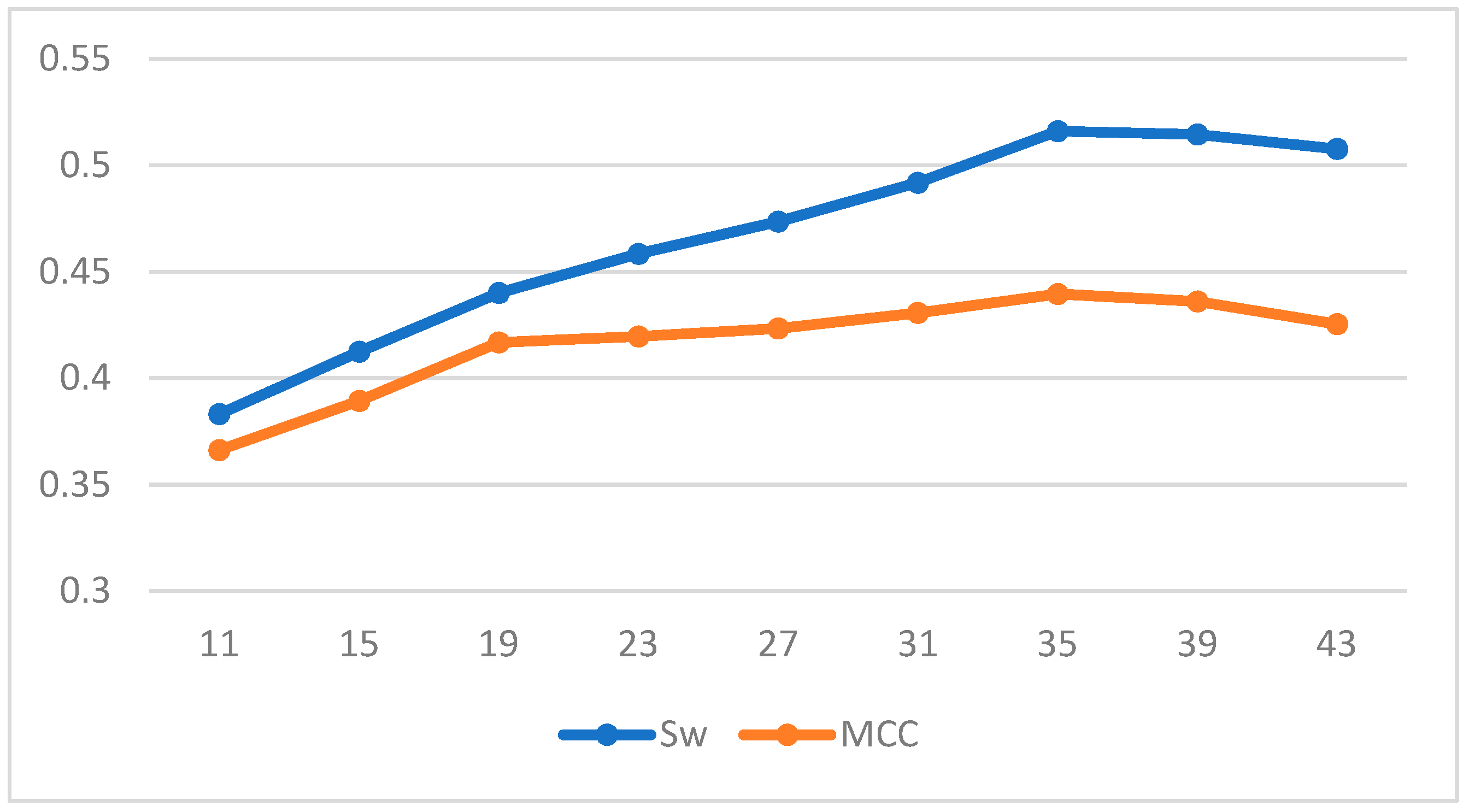
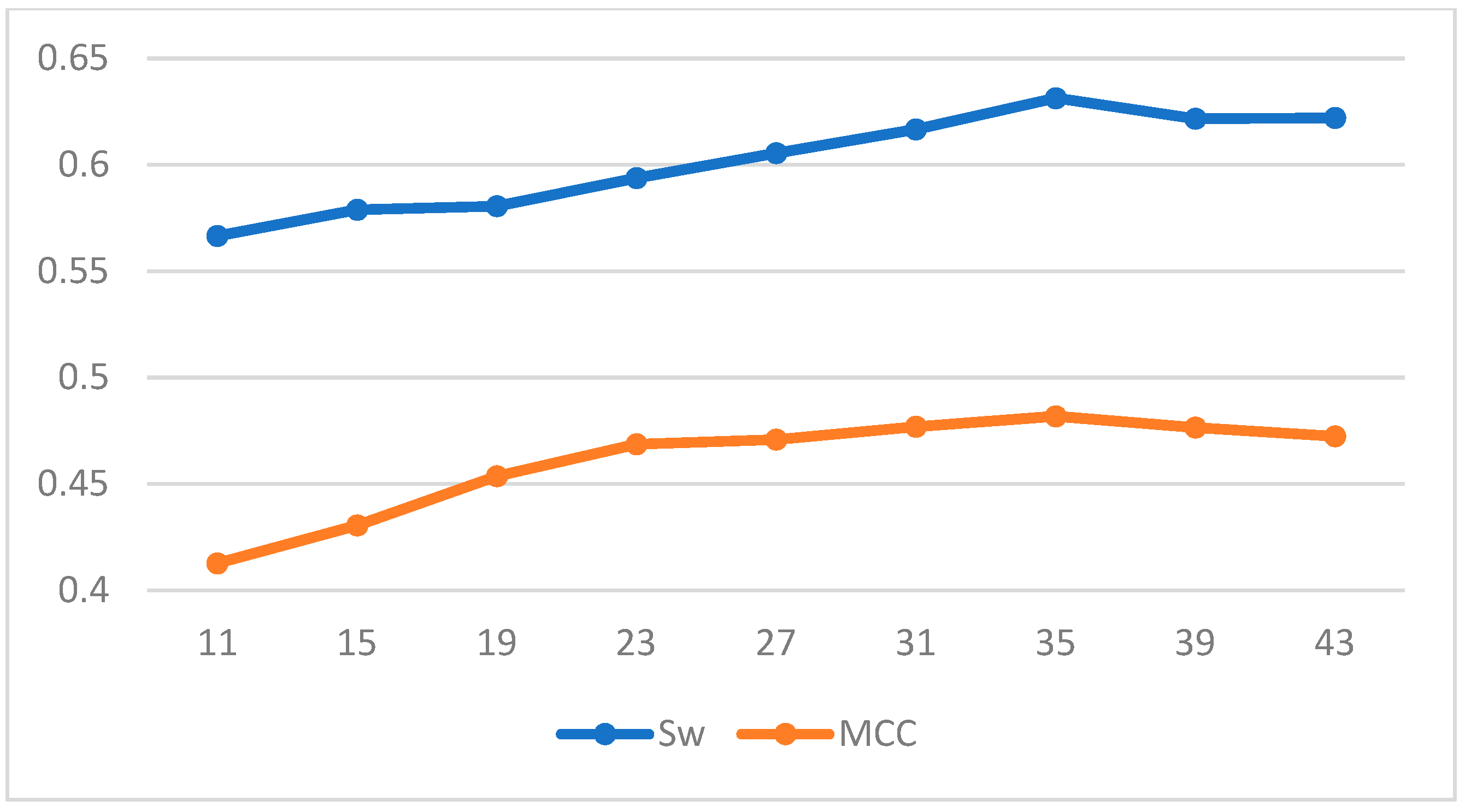
| Length | LDA | SVM | ||
|---|---|---|---|---|
| Sw | MCC | Sw | MCC | |
| 11 | 0.3831 | 0.3662 | 0.5665 | 0.4127 |
| 15 | 0.4125 | 0.3893 | 0.5788 | 0.4305 |
| 19 | 0.4401 | 0.4168 | 0.5805 | 0.4536 |
| 23 | 0.4584 | 0.4196 | 0.5937 | 0.4686 |
| 27 | 0.4736 | 0.4233 | 0.6054 | 0.4708 |
| 31 | 0.4918 | 0.4307 | 0.6166 | 0.4769 |
| 35 | 0.5161 | 0.4396 | 0.6313 | 0.4818 |
| 39 | 0.5145 | 0.4361 | 0.6216 | 0.4765 |
| 43 | 0.5078 | 0.4254 | 0.6220 | 0.4723 |
| 47 | 0.4854 | 0.4260 | 0.6139 | 0.4668 |
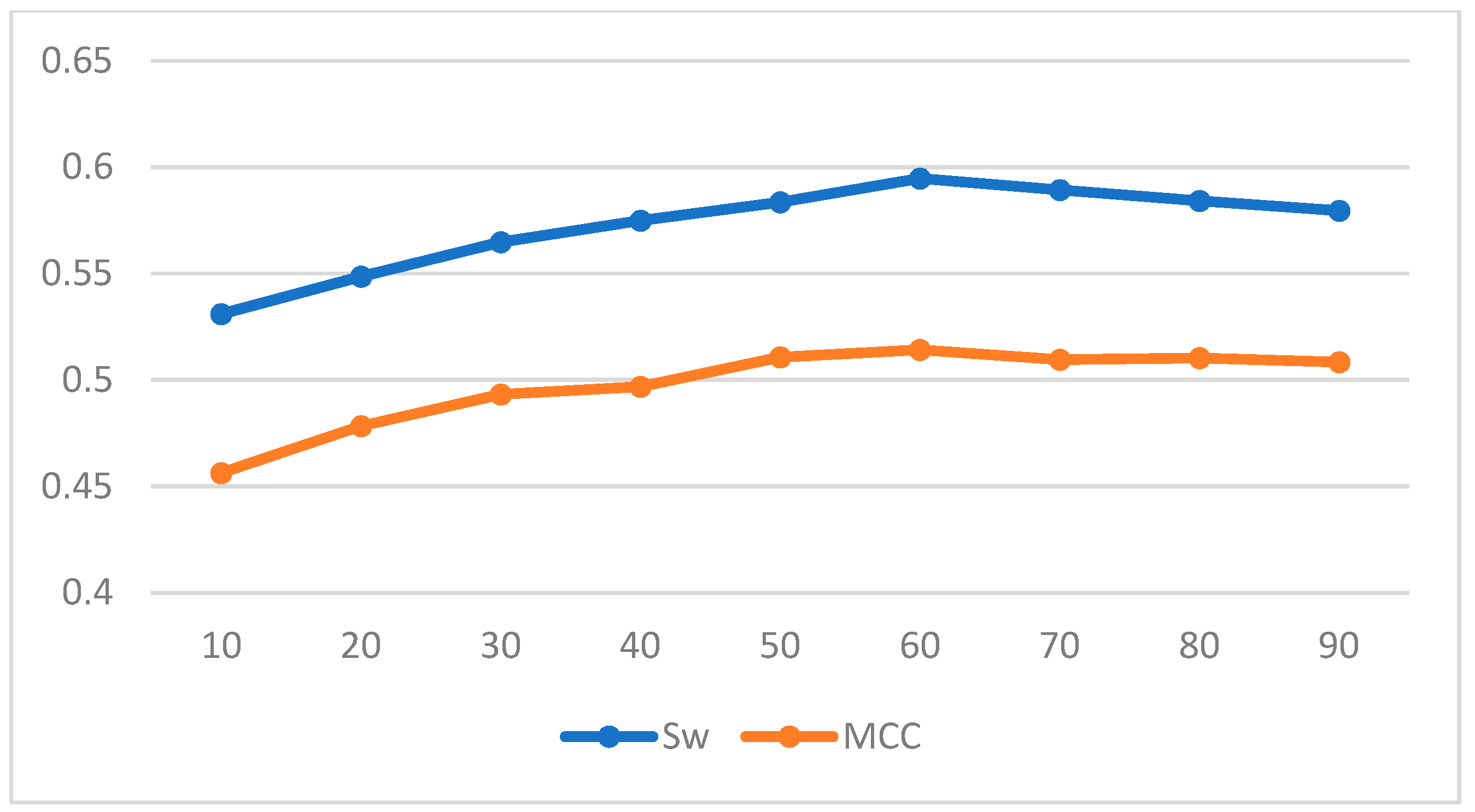
| Length | Sw | MCC |
|---|---|---|
| 10 | 0.5310 | 0.4563 |
| 20 | 0.5486 | 0.4783 |
| 30 | 0.5648 | 0.4933 |
| 40 | 0.5749 | 0.4968 |
| 50 | 0.5836 | 0.5107 |
| 60 | 0.5947 | 0.5142 |
| 70 | 0.5893 | 0.5096 |
| 80 | 0.5842 | 0.5103 |
| 90 | 0.5796 | 0.5084 |
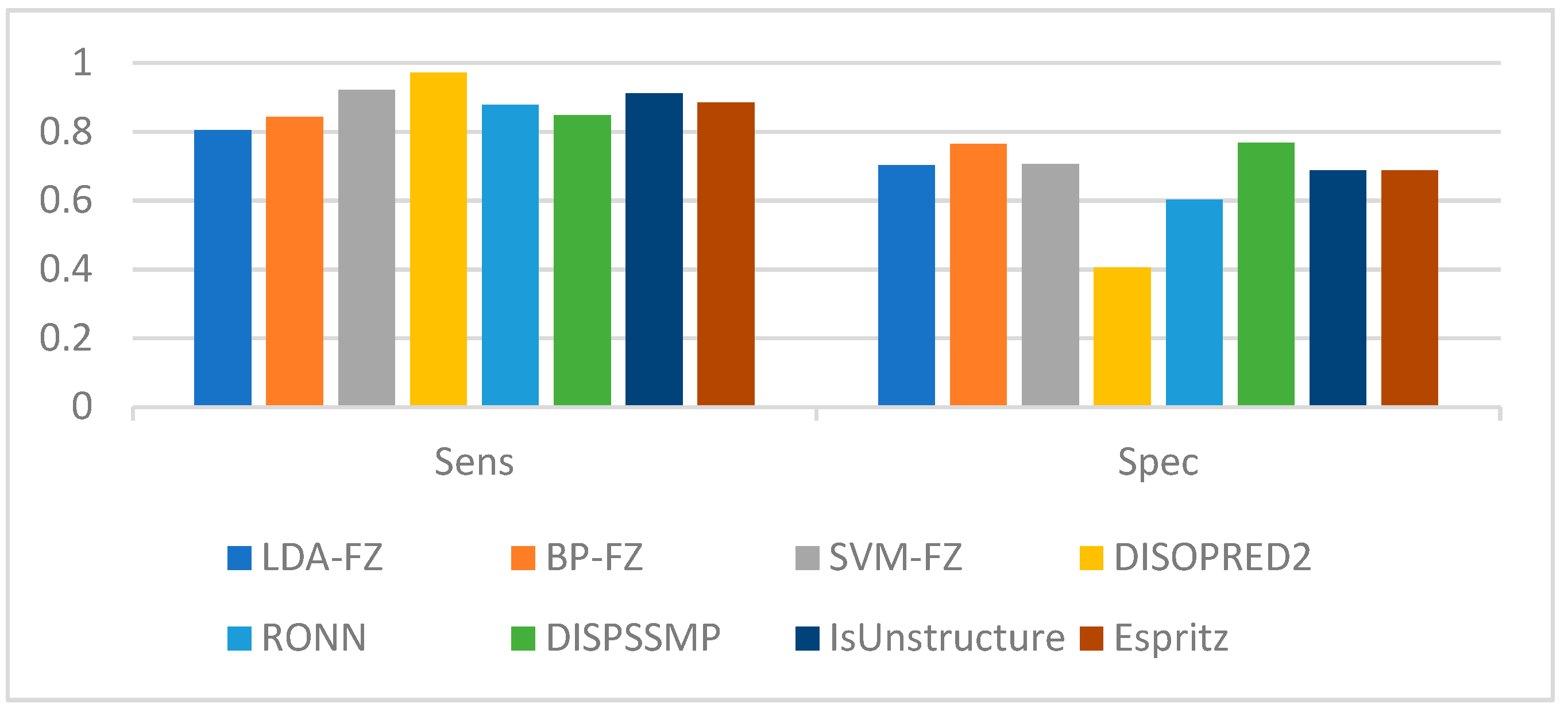
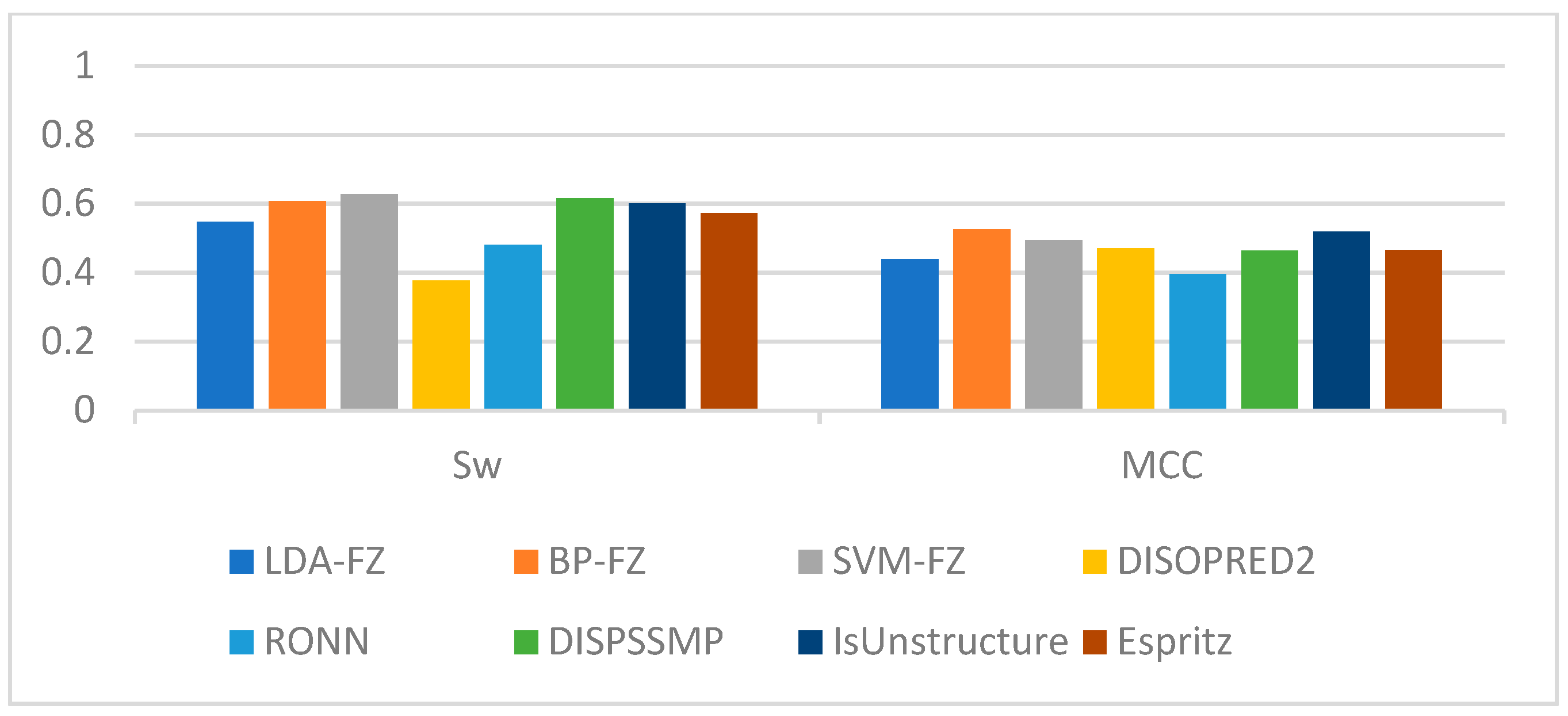
| Sens | Spec | Sw | MCC | |
|---|---|---|---|---|
| LDA-FE | 0.846 | 0.702 | 0.548 | 0.439 |
| BP-FE | 0.843 | 0.765 | 0.608 | 0.518 |
| SVM-FE | 0.921 | 0.706 | 0.627 | 0.493 |
| DISOPRED2 | 0.972 | 0.405 | 0.377 | 0.470 |
| RONN | 0.878 | 0.603 | 0.481 | 0.395 |
| DISPSSMP | 0.848 | 0.767 | 0.615 | 0.463 |
| IsUnstructure | 0.911 | 0.688 | 0.600 | 0.518 |
| Espritz | 0.884 | 0.688 | 0.572 | 0.466 |
| Sens | Spec | Sw | MCC | |
|---|---|---|---|---|
| Fuzzy entropy included | 0.8461 | 0.7023 | 0.5484 | 0.4396 |
| Fuzzy entropy not included | 0.8263 | 0.6902 | 0.5165 | 0.4218 |
| Sens | Spec | Sw | MCC | |
|---|---|---|---|---|
| Fuzzy entropy included | 0.9213 | 0.7061 | 0.6394 | 0.4932 |
| Fuzzy entropy not included | 0.9016 | 0.6768 | 0.5784 | 0.4746 |
| Sens | Spec | Sw | MCC | |
|---|---|---|---|---|
| Fuzzy entropy included | 0.8431 | 0.7654 | 0.6085 | 0.5184 |
| Fuzzy entropy not included | 0.8243 | 0.7548 | 0.5771 | 0.4826 |
| SVM-FE & BP-FE | SVM-FE & LDA-FE | BP-FE & LDA-FE | |
|---|---|---|---|
| p-value | 0.1679 | 9.5886 × 10−10 | 2.1404 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Liu, H.; He, H. Prediction of Intrinsically Disordered Proteins Using Machine Learning Algorithms Based on Fuzzy Entropy Feature. Algorithms 2021, 14, 102. https://doi.org/10.3390/a14040102
Zhang L, Liu H, He H. Prediction of Intrinsically Disordered Proteins Using Machine Learning Algorithms Based on Fuzzy Entropy Feature. Algorithms. 2021; 14(4):102. https://doi.org/10.3390/a14040102
Chicago/Turabian StyleZhang, Lin, Haiyuan Liu, and Hao He. 2021. "Prediction of Intrinsically Disordered Proteins Using Machine Learning Algorithms Based on Fuzzy Entropy Feature" Algorithms 14, no. 4: 102. https://doi.org/10.3390/a14040102
APA StyleZhang, L., Liu, H., & He, H. (2021). Prediction of Intrinsically Disordered Proteins Using Machine Learning Algorithms Based on Fuzzy Entropy Feature. Algorithms, 14(4), 102. https://doi.org/10.3390/a14040102