
Classification of Precursor MicroRNAs from Different Species Based on K-mer Distance Features



Abstract
:1. Background
2. Methods
2.1. Feature Space of Precursor miRNA
2.2. K-mer Features
2.3. K-mer Distance Features
2.3.1. Inter K-mer Distance
2.3.2. K-mer First–Last Distance
2.3.3. K-mer Location Distance
2.4. Secondary Features
2.5. Other Features Describing Pre-miRNAs
3. Datasets and Methods
Preprocessing the Data
4. Feature Vector and Feature Selection
5. Classification Approach
6. Model Performance Evaluation
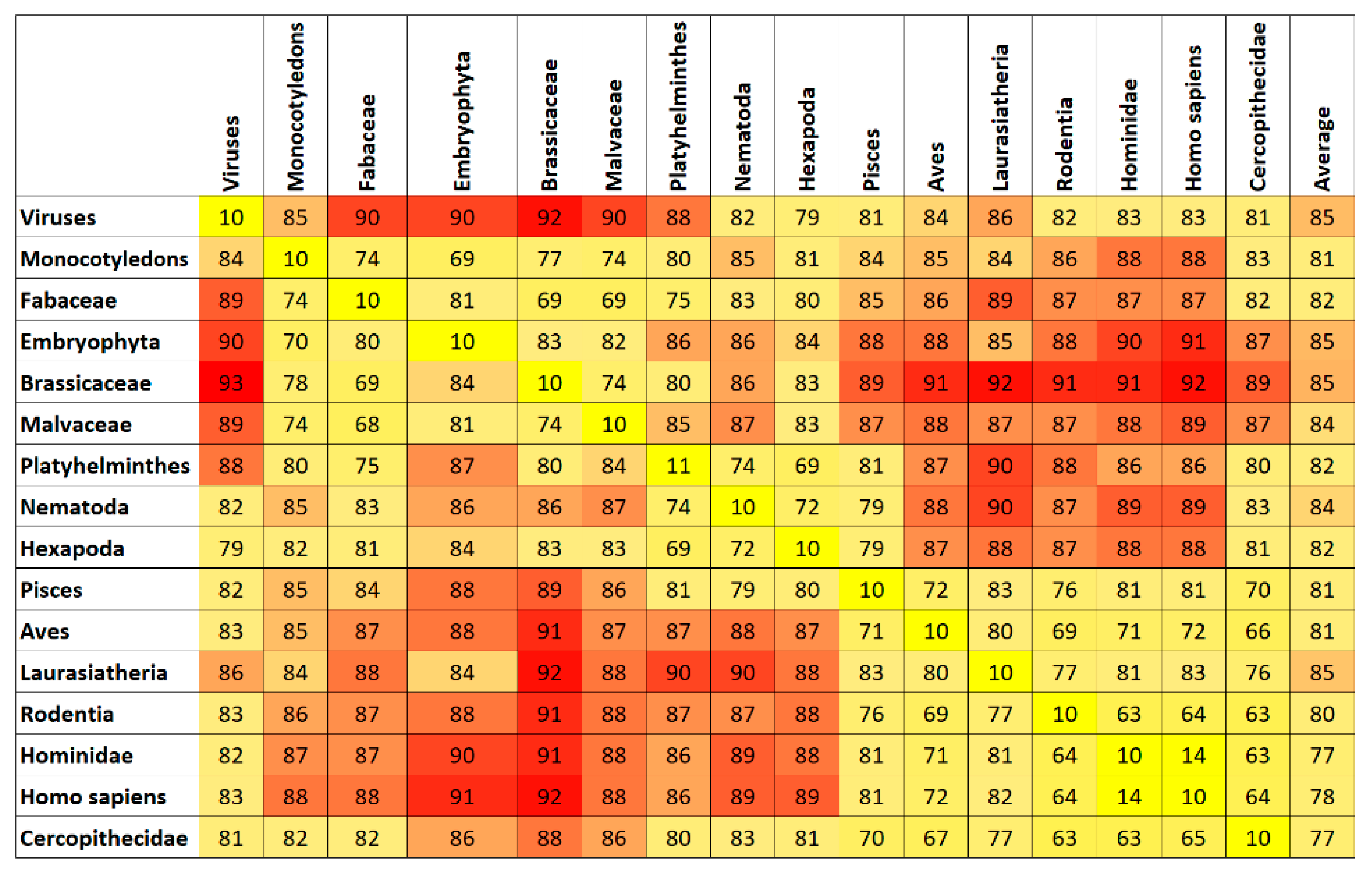
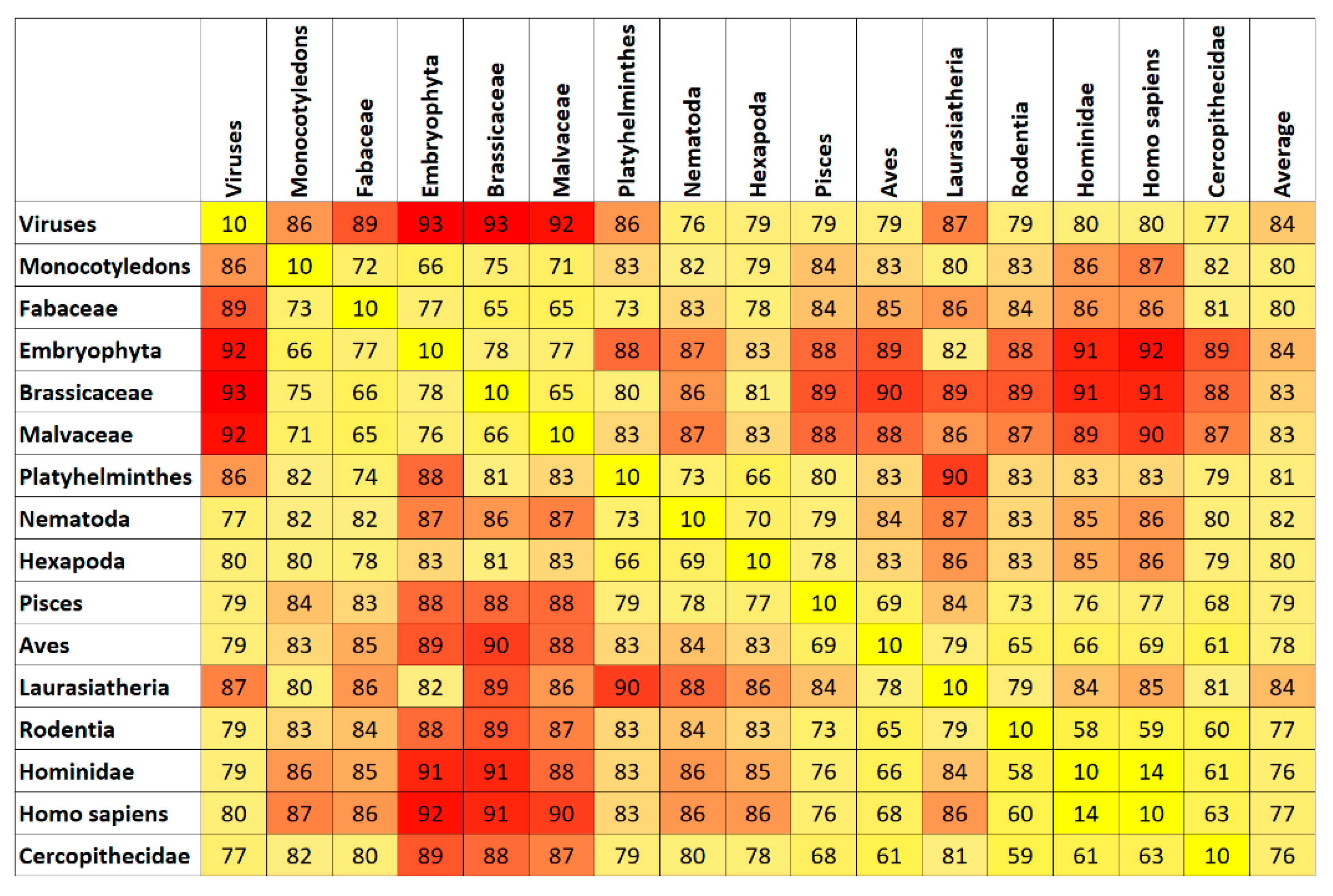
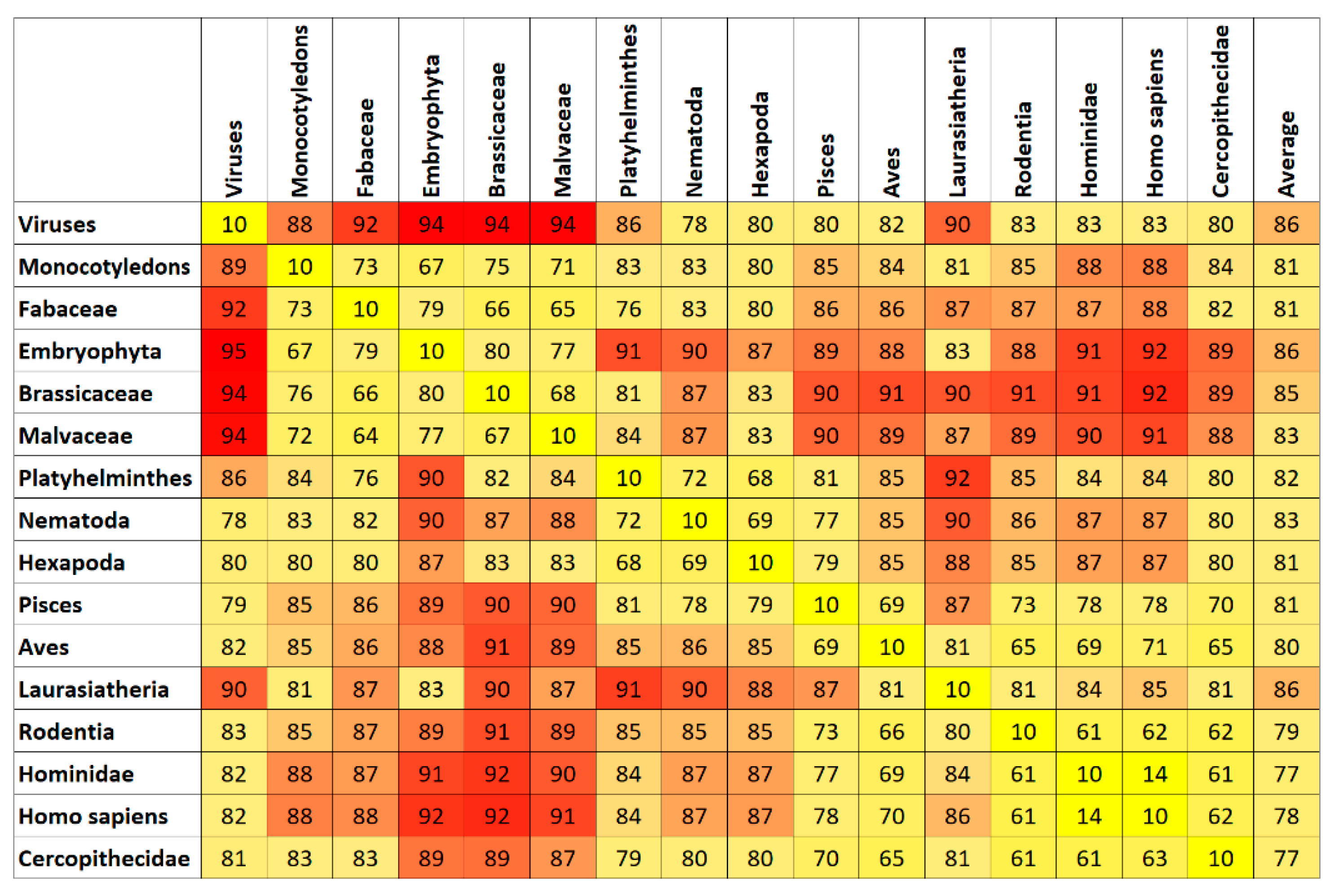
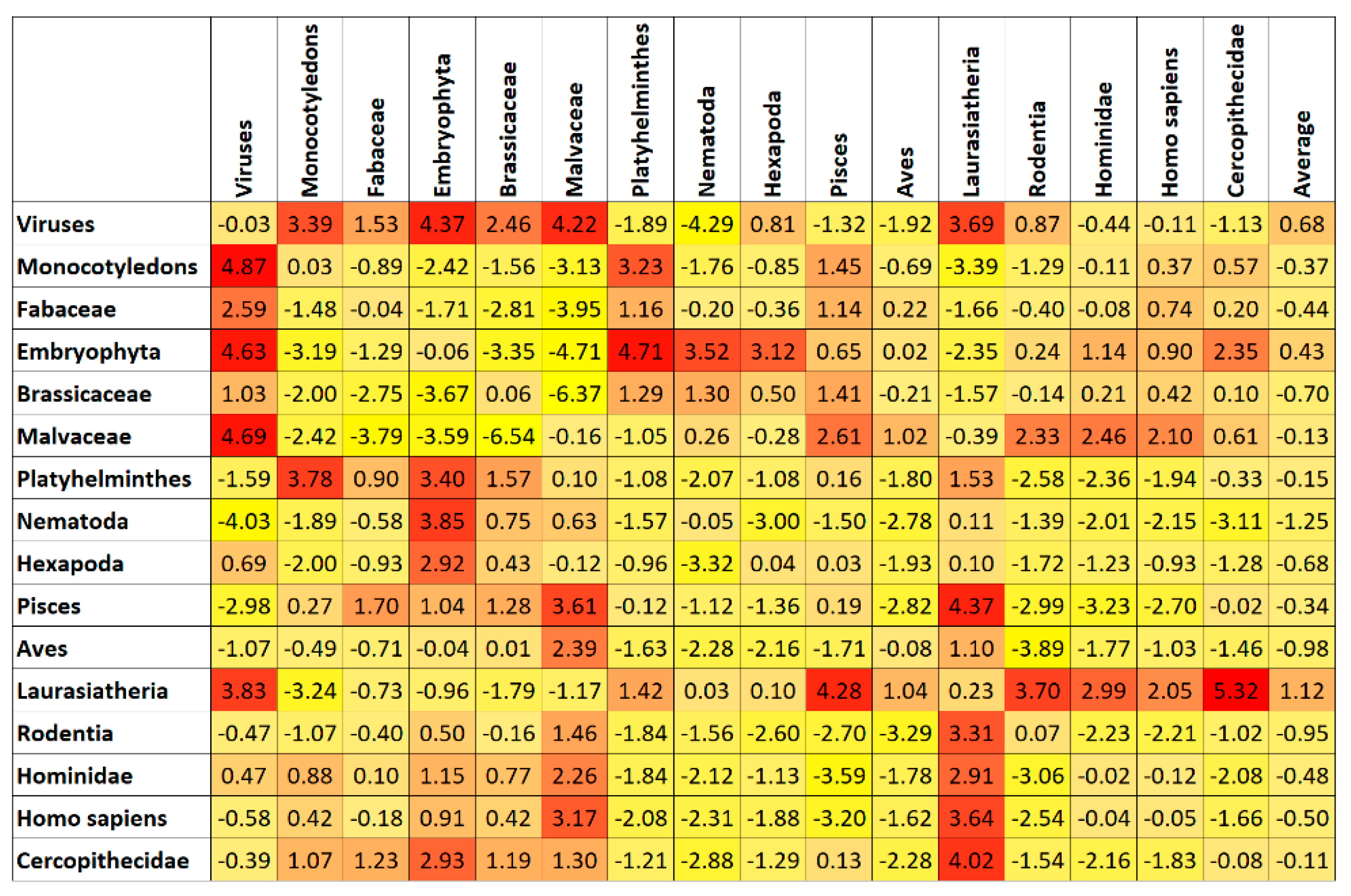
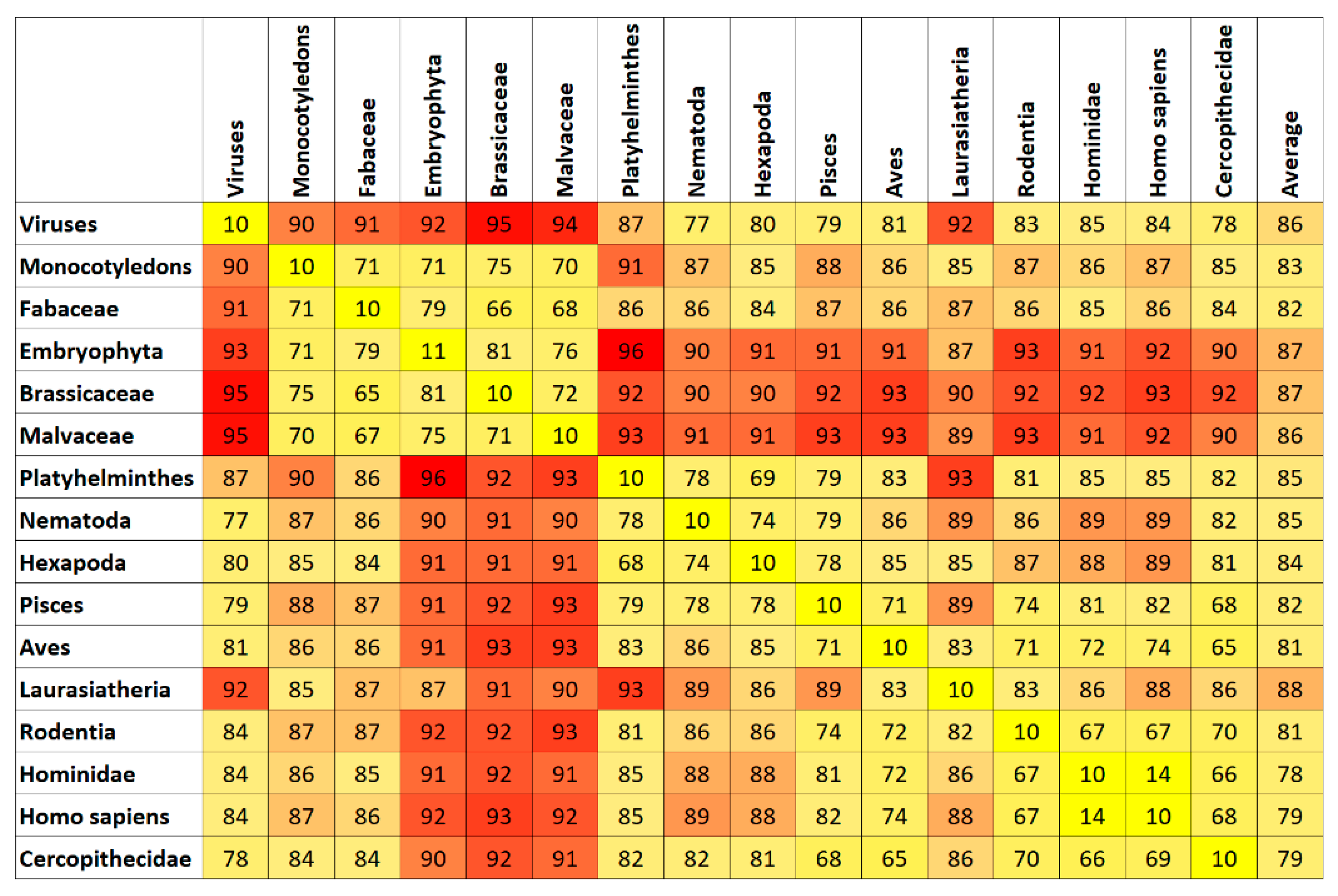
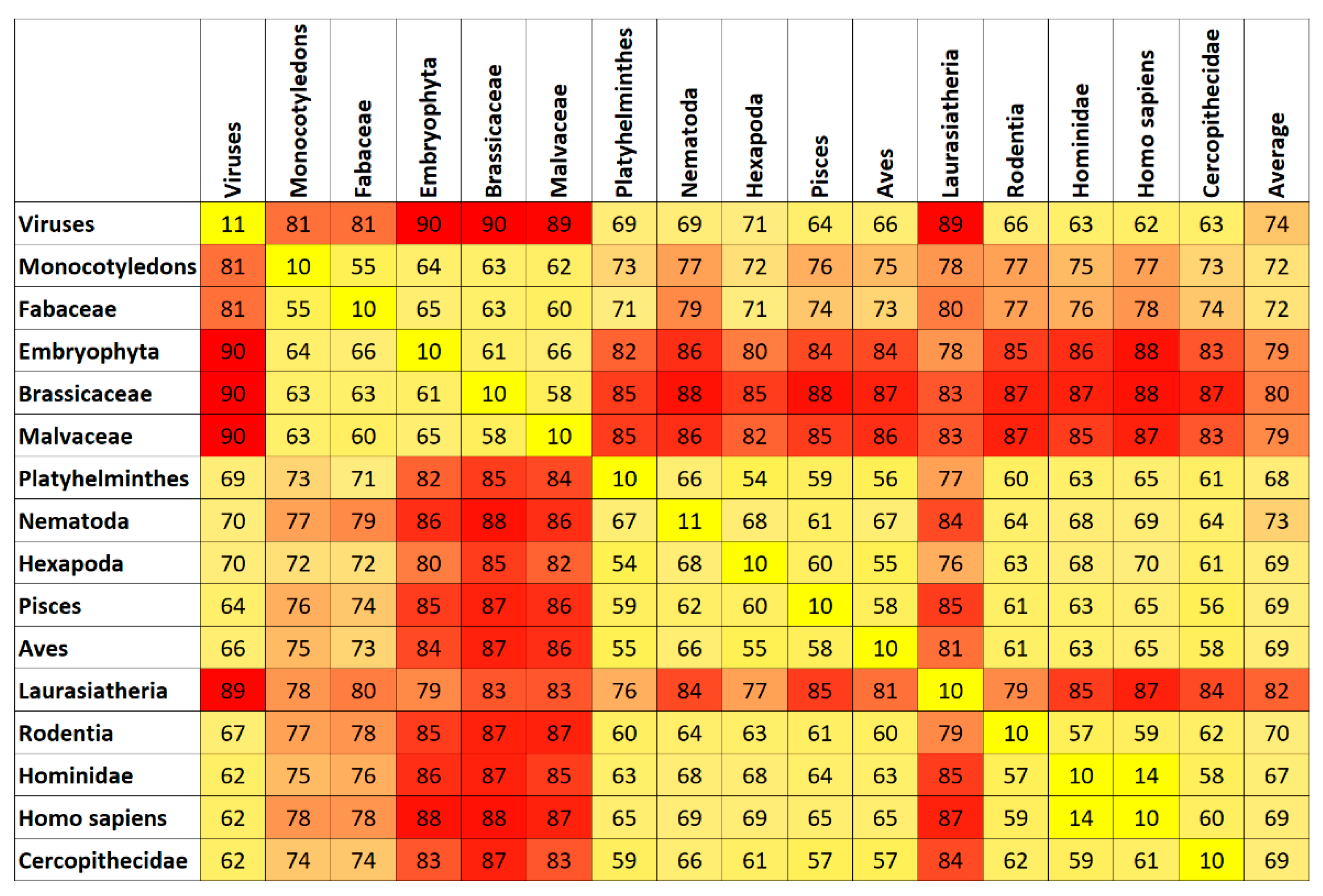
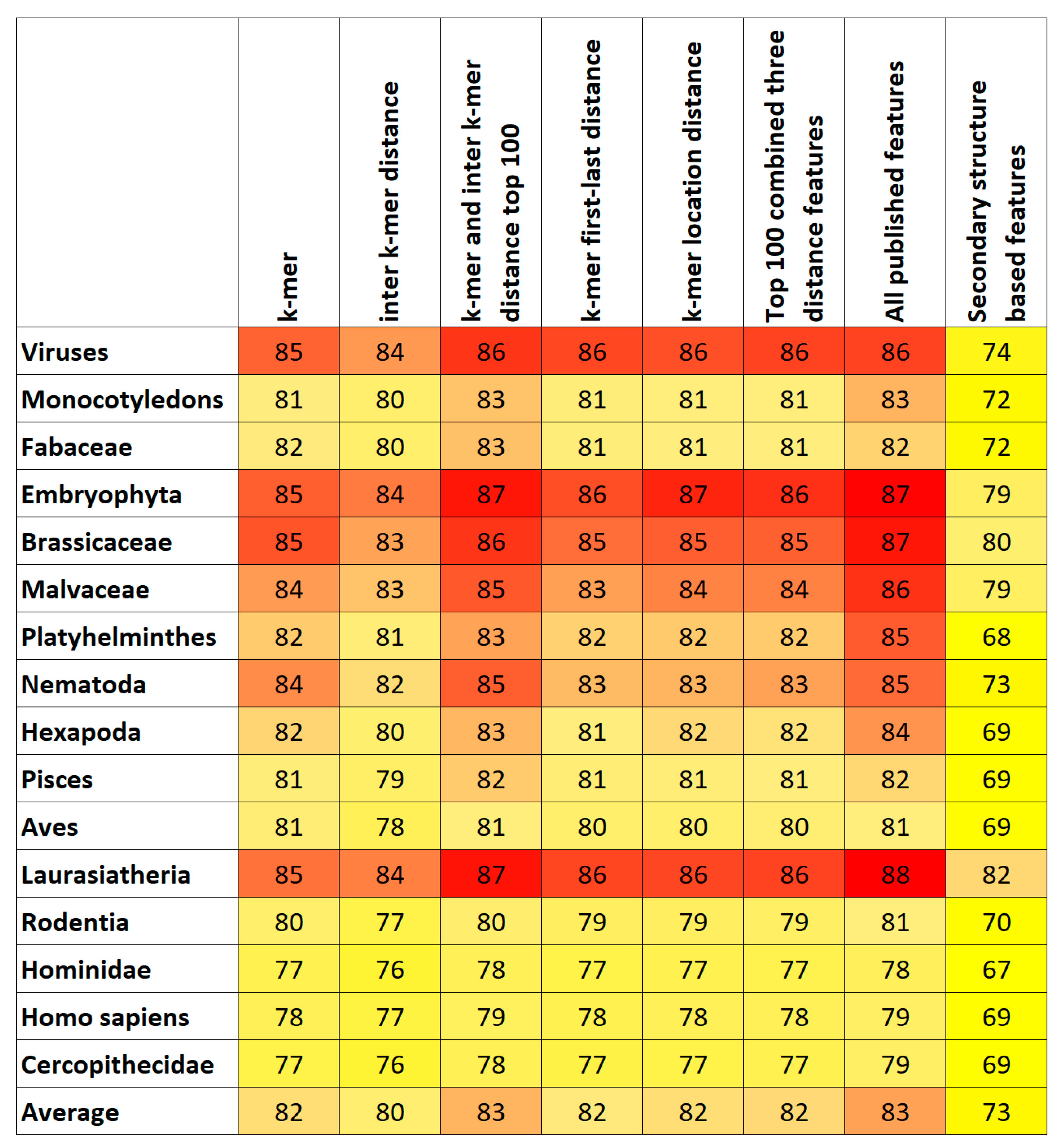
7. Results and Discussion
Top Features
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Availability and Implementation
References
- Erson-Bensan, A.E. Introduction to MicroRNAs in Biological Systems. Methods Mol. Biol. 2014, 1107, 1–14. [Google Scholar] [CrossRef]
- Chapman, E.J.; Carrington, J.C. Specialization and Evolution of Endogenous Small RNA Pathways. Nat. Rev. Genet. 2007, 8, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Allmer, J.; Khalifa, W. Plant MicroRNA Prediction Employing Sequence Motifs Achieves High Accuracy. 2016. Available online: https://www.researchgate.net/publication/320402782_Plant_microRNA_prediction_employing_sequence_motifs_achieves_high_accuracy (accessed on 21 April 2021).
- Grey, F. Role of MicroRNAs in Herpesvirus Latency and Persistence. J. Gen. Virol. 2015, 96, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Saçar, M.D.; Allmer, J. Current Limitations for Computational Analysis of MiRNAs in Cancer. Pak. J. Clin. Biomed. Res. 2013, 1, 3–5. [Google Scholar]
- Yousef, M.; Trinh, H.V.; Allmer, J. Intersection of MicroRNA and Gene Regulatory Networks and Their Implication in Cancer. Curr. Pharm. Biotechnol. 2014, 15, 445–454. [Google Scholar] [CrossRef]
- Allmer, J.; Yousef, M. Computational Methods for Ab Initio Detection of MicroRNAs. Front. Genet. 2012, 3, 209. [Google Scholar] [CrossRef] [Green Version]
- Saçar, M.; Allmer, J. Machine Learning Methods for MicroRNA Gene Prediction. In miRNomics: MicroRNA Biology and Computational Analysis SE-10; Yousef, M., Allmer, J., Eds.; Methods in Molecular Biology; Humana Press: Tortowa, NJ, USA, 2014; Volume 1107, pp. 177–187. ISBN 978-1-62703-747-1. [Google Scholar]
- Yousef, M.; Nebozhyn, M.; Shatkay, H.; Kanterakis, S.; Showe, L.C.; Showe, M.K. Combining Multi-Species Genomic Data for MicroRNA Identification Using a Naive Bayes Classifier. Bioinformatics 2006, 22, 1325–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, H.T.; Tho, H.P.; Satou, K.; Tu, B.H. Prediction of MicroRNA Hairpins Using One-Class Support Vector Machines. In Proceedings of the 2nd International Conference on Bioinformatics and Biomedical Engineering, iCBBE 2008, Shanghai, China, 16–18 May 2008; pp. 33–36. [Google Scholar]
- Khalifa, W.; Yousef, M.; Demirci, M.D.S.; Allmer, J. The Impact of Feature Selection on One and Two-Class Classification Performance for Plant MicroRNAs. PeerJ 2016, 4, e2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, M.; Jung, S.; Showe, L.C.; Showe, M.K. Learning from Positive Examples When the Negative Class Is Undetermined—MicroRNA Gene Identification. Algorithms Mol. Biol. AMB 2008, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Demirci, M.D.S.; Allmer, J. Delineating the Impact of Machine Learning Elements in Pre-MicroRNA Detection. PeerJ 2017, 5, e3131. [Google Scholar] [CrossRef] [Green Version]
- Saçar, M.D.; Hamzeiy, H.; Allmer, J. Can MiRBase Provide Positive Data for Machine Learning for the Detection of MiRNA Hairpins? J. Integr. Bioinform. 2013, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate MicroRNA Genes and the Evolution of the Human MicroRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duygu, M.; Demirci, S.; Allmer, J. Improving the Quality of Positive Datasets for the Establishment of Machine Learning Models for Pre- MicroRNA Detection. J. Integr. Bioinform. 2017, 14, 20170032. [Google Scholar]
- Hsu, S.-D.; Tseng, Y.-T.; Shrestha, S.; Lin, Y.-L.; Khaleel, A.; Chou, C.-H.; Chu, C.-F.; Huang, H.-Y.; Lin, C.-M.; Ho, S.-Y.; et al. MiRTarBase Update 2014: An Information Resource for Experimentally Validated MiRNA-Target Interactions. Nucleic Acids Res. 2014, 42, D78–D85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergoulis, T.; Vlachos, I.S.; Alexiou, P.; Georgakilas, G.; Maragkakis, M.; Reczko, M.; Gerangelos, S.; Koziris, N.; Dalamagas, T.; Hatzigeorgiou, A.G. TarBase 6.0: Capturing the Exponential Growth of MiRNA Targets with Experimental Support. Nucleic Acids Res. 2012, 40, D222–D229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating MicroRNA Annotation and Deep-Sequencing Data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirci, M.D.S.; Baumbach, J.; Allmer, J. On the Performance of Pre-MicroRNA Detection Algorithms. Nat. Commun. 2017, 8, 330. [Google Scholar] [CrossRef]
- Sacar, M.D.; Allmer, J. Data Mining for Microrna Gene Prediction: On the Impact of Class Imbalance and Feature Number for Microrna Gene Prediction. In Proceedings of the 2013 8th International Symposium on Health Informatics and Bioinformatics, Ankara, Turkey, 25–27 September 2013. [Google Scholar]
- Sewer, A.; Paul, N.; Landgraf, P.; Aravin, A.; Pfeffer, S.; Brownstein, M.J.; Tuschl, T.; van Nimwegen, E.; Zavolan, M. Identification of Clustered MicroRNAs Using an Ab Initio Prediction Method. BMC Bioinform. 2005, 6, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Sobczak, K.; Wilczynska, U.; Drath, M.; Jasinska, A.; Kaczynska, D.; Krzyzosiak, W.J. Structural Features of MicroRNA (MiRNA) Precursors and Their Relevance to MiRNA Biogenesis and Small Interfering RNA/Short Hairpin RNA Design. J. Biol. Chem. 2004, 279, 42230–42239. [Google Scholar] [CrossRef] [Green Version]
- Yones, C.A.; Stegmayer, G.; Kamenetzky, L.; Milone, D.H. MiRNAfe: A Comprehensive Tool for Feature Extraction in MicroRNA Prediction. BioSystems 2015, 138, 1–5. [Google Scholar] [CrossRef]
- Lai, E.C.; Tomancak, P.; Williams, R.W.; Rubin, G.M. Computational Identification of Drosophila MicroRNA Genes. Genome Biol. 2003, 4, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, M.; Khalifa, W.; Acar, I.E.; Allmer, J. MicroRNA Categorization Using Sequence Motifs and K-Mers. BMC Bioinform. 2017, 18, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, M.; Nigatu, D.; Levy, D.; Allmer, J.; Henkel, W. Categorization of Species Based on Their MicroRNAs Employing Sequence Motifs, Infor-Mation-Theoretic Sequence Feature Extraction, and k-Mers. EURASIP J. Adv. Signal Process. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Cakir, M.V.; Allmer, J. Systematic Computational Analysis of Potential RNAi Regulation in Toxoplasma Gondii. In Proceedings of the 2010 5th International Symposium on Health Informatics and Bioinformatics, Ankara, Turkey, 20–22 April 2010. [Google Scholar]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. Available online: https://www.knime.com/sites/default/files/knime_whitepaper.pdf (accessed on 21 April 2021).
- Griffiths-Jones, S. MiRBase: MicroRNA Sequences and Annotation. Curr. Protoc. Bioinform. 2010, 29, 12.9.1–12.9.10. [Google Scholar] [CrossRef]
- Ng, K.L.S.; Mishra, S.K. De Novo SVM Classification of Precursor MicroRNAs from Genomic Pseudo Hairpins Using Global and Intrinsic Folding Measures. Bioinformatics 2007, 23, 1321–1330. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, W.; Gao, D.; Rasko, J.E.J. Defining and Providing Robust Controls for MicroRNA Prediction. Bioinformatics 2012, 28, 1058–1061. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Wu, H.; Wang, W.; Ma, W.; Sun, X.; Lu, Z. MiPred: Classification of Real and Pseudo MicroRNA Precursors Using Random Forest Prediction Model with Combined Features. Nucleic Acids Res. 2007, 35, W339–W344. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Li, F.; He, T.; Liu, G.-P.; Li, Y.; Zhang, X. Classification of Real and Pseudo MicroRNA Precursors Using Local Structure-Sequence Features and Support Vector Machine. BMC Bioinform. 2005, 6, 310. [Google Scholar] [CrossRef] [Green Version]
- Yousef, M.; Allmer, J.; Khalifa, W. Sequence Motif-Based One-Class Classifiers Can Achieve Comparable Accuracy to Two-Class Learners for Plant MicroRNA Detection. J. Biomed. Sci. Eng. 2015. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.-S.; Liang, Y.-Z. Monte Carlo Cross Validation. Chemom. Intell. Lab. Syst. 2001, 56, 1–11. [Google Scholar] [CrossRef]
- Matthews, B.W. Comparison of the Predicted and Observed Secondary Structure of T4 Phage Lysozyme. BBA Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Saçar Demirci, M.D.; Bağci, C.; Allmer, J. Differential Expression of Toxoplasma Gondii MicroRNAs in Murine and Human Hosts. Available online: https://openaccess.iyte.edu.tr/xmlui/bitstream/handle/11147/7918/[email protected];jsessionid=D7A7AB90CE83A13466B77615F319E128?sequence=1 (accessed on 21 April 2021).
- Saçar, M.D.; Bağcı, C.; Allmer, J. Computational Prediction of MicroRNAs from Toxoplasma Gondii Potentially Regulating the Hosts’ Gene Expression. Genom. Proteom. Bioinform. 2014, 12, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzer, A.; Stadler, P.F. Evolution of MicroRNAs. Methods Mol. Biol. 2006, 342, 335–350. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| #Precursors | #Uniques | |
|---|---|---|
| Hominidae | 3629 | 1326 |
| Brassicaceae | 726 | 535 |
| Hexapoda | 3119 | 2050 |
| Monocotyledons (Liliopsida) | 1598 | 1402 |
| Nematoda | 1789 | 1632 |
| Fabaceae | 1313 | 1011 |
| Pisces (Chondricthyes) | 1530 | 682 |
| Virus | 306 | 295 |
| Aves | 948 | 790 |
| Laurasiatheria | 1205 | 675 |
| Rodentia | 1778 | 993 |
| Homo sapiens | 1828 | 1223 |
| Cercopithecidae | 631 | 503 |
| Embryophyta | 287 | 278 |
| Malvaceae | 458 | 419 |
| Platyhelminthes | 424 | 381 |
| Total | 21,569 | 14,195 |
| Aves vs. Brassicaceae | Hexapoda vs. Embryophyta | ||
|---|---|---|---|
| Feature | IG | Feature | IG |
| AU_all | 0.45 | G_all | 0.36 |
| U_all | 0.43 | U_all | 0.36 |
| A_all | 0.42 | A_all | 0.34 |
| UU_all | 0.41 | UG_all | 0.33 |
| UA_all | 0.41 | C_all | 0.33 |
| AA_all | 0.39 | CU_all | 0.30 |
| AUU_all | 0.39 | AG_all | 0.29 |
| AAU_all | 0.37 | CA_all | 0.29 |
| G_all | 0.37 | GC_all | 0.28 |
| AUA_all | 0.35 | UU_all | 0.27 |
| UAU_all | 0.34 | GA_all | 0.26 |
| AU_AU | 0.34 | AU_all | 0.25 |
| GA_all | 0.33 | GU_all | 0.24 |
| AUU_AUU | 0.33 | UC_all | 0.23 |
| UUU_UUU | 0.32 | AC_all | 0.22 |
| UU_UU | 0.32 | GG_all | 0.21 |
| C_all | 0.32 | UGC_all | 0.20 |
| UA_UA | 0.32 | CC_all | 0.19 |
| AAU_AAU | 0.31 | UA_all | 0.19 |
| GU_all | 0.31 | AA_all | 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousef, M.; Allmer, J. Classification of Precursor MicroRNAs from Different Species Based on K-mer Distance Features. Algorithms 2021, 14, 132. https://doi.org/10.3390/a14050132
Yousef M, Allmer J. Classification of Precursor MicroRNAs from Different Species Based on K-mer Distance Features. Algorithms. 2021; 14(5):132. https://doi.org/10.3390/a14050132
Chicago/Turabian StyleYousef, Malik, and Jens Allmer. 2021. "Classification of Precursor MicroRNAs from Different Species Based on K-mer Distance Features" Algorithms 14, no. 5: 132. https://doi.org/10.3390/a14050132
APA StyleYousef, M., & Allmer, J. (2021). Classification of Precursor MicroRNAs from Different Species Based on K-mer Distance Features. Algorithms, 14(5), 132. https://doi.org/10.3390/a14050132