
Predicting the Evolution of Syntenies—An Algorithmic Review

{kind=link}
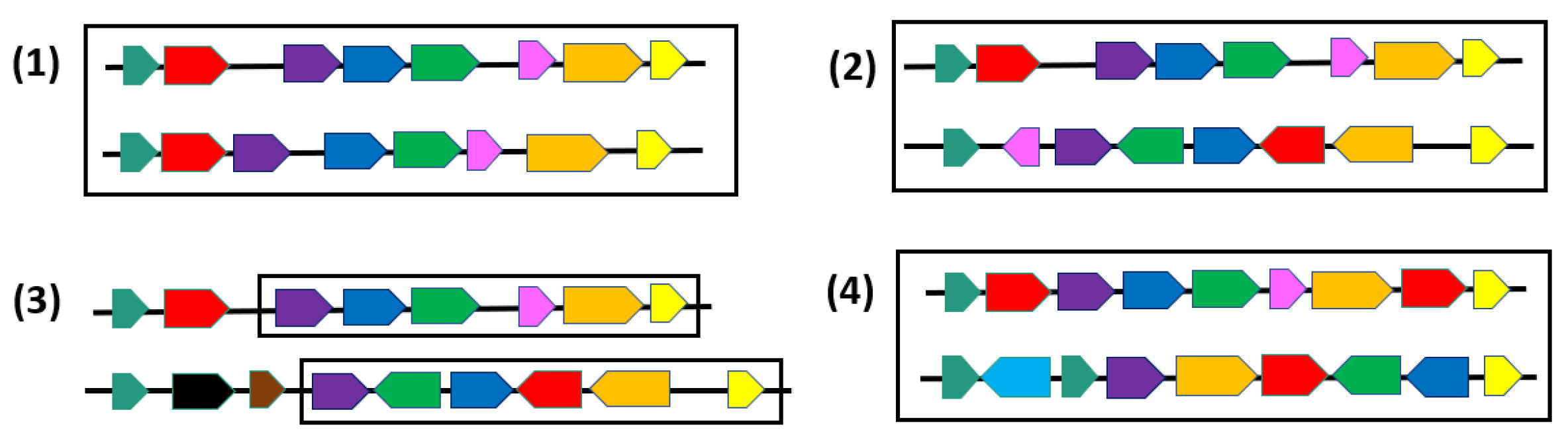{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Syntenies Defined as Gene Orders
2.1. Gene Families
2.2. Trees
3. The Sorting by Rearrangement Problem
The Small Phylogeny Problem
| Algorithm 1 Small Phylogeny Problem |
| Small Phylogeny Problem: |
| Input: A phylogenetic tree S for a set of species, a set of gene families, a set X of syntenies on labeling the leaves of S and a model of evolution; |
| Output: A synteny labeling of the internal nodes of S minimizing the total branch length over the phylogeny. |
4. Accounting for Gene Gain and Loss
5. Accounting for Gene Trees
5.1. The Reconciliation Approach
- 1.
- and are the two children of in S in which case ;
- 2.
- in which case representing a duplication in σ;
5.2. Adjacency Evolution
5.3. Evolution through Segmental Duplications and Losses
- a speciation acting on a synteny belonging to a genome has the effect of reproducing X in the two genomes and children of in S;
- a (segmental) duplication acting on a synteny X belonging to a genome is an operation that copies a substring of somewhere else into the genome , creating a new copied synteny, where each , for , belongs to the same gene family as ;
- a (segmental) loss acting on a is an operation that removes a substring of X, leading to the truncated synteny . A loss is called full if is the empty string (i.e., all genes of X are removed) and partial otherwise. A partial loss event is denoted .
- Given the set of gene trees for , find a synteny tree T for ;
- Given a species tree S for , find a Super-reconciliation of T with S, i.e., an event-labeled synteny tree which is a “partial extension” of T, representing a valid history for , of minimum DL-distance. Here, the DL-distance of is the number of induced segmental duplications and losses.
5.3.1. Synteny Tree Reconstruction
5.3.2. Super-Reconciliation
5.3.3. Unordered Super-Reconciliation
- , then x is a binary node with two children corresponding to syntenies Y and Z such that and and are the two children of in S.
- , then x is a binary node with two children corresponding to syntenies Y and Z such that , and .
- , then x is a unary node with a child corresponding to a synteny Y such that and .
5.4. Minimizing Duplication Episodes
5.4.1. The Interval Model
5.4.2. The Clustering Model
5.5. Evolution of Tandemly Arrayed Gene Clusters
5.6. Accounting for Horizontal Gene Transfers
6. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tremblay-Savard, O.; Benzaid, B.; Lang, B.F.; El-Mabrouk, N. Evolution of tRNA Repertoires in Bacillus Inferred with OrthoAlign. Mol. Biol. Evol. 2015, 32, 1643–1656. [Google Scholar] [CrossRef] [Green Version]
- Goodman, M.; Czelusniak, J.; Moore, G.; Romero-Herrera, A.; Matsuda, G. Fitting the gene lineage into its species lineage, a parsimony strategy illustrated by cladograms constructed from globin sequences. Syst. Zool. 1979, 28, 132–163. [Google Scholar] [CrossRef]
- Larsson, T.; Olsson, F.; Sundstrom, G.; Lundin, L.; Brenner, S.; Venkatesh, B.; Larhammar, D. Early vertebrate chromosome duplications and the evolution of the neuropeptide Y receptor gene regions. BMC Evol. Biol. 2008, 8, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, A.; Grzeschik, K. An insight into the phylogenetic history of HOX linked gene families in vertebrates. BMC Evol. Biol. 2007, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrier, D. Evolution of Homeobox Gene Clusters in Animals:The Giga-Cluster and Primary vs. Secondary Clustering. Front. Ecol. Evol. 2016, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Fernàndez, J. The genesis and evolution of Homeobox gene clusters. Nat. Rev. Genet. 2005, 6, 881–892. [Google Scholar] [CrossRef]
- Ajmal, W.; Khan, H.; Abbasi, A. Phylogenetic investigation of human FGFR-bearing paralogons favors piecemeal duplication theory of vertebrate genome evolution. Mol. Phylogenet. Evol. 2014, 81, 49–60. [Google Scholar] [CrossRef]
- Hafeez, M.; Shabbir, M.; Altaf, F.; Abbasi, A. Phylogenomic analysis reveals ancient segmental duplications in the human genome. Mol. Phylogenet. Evol. 2016, 94, 95–100. [Google Scholar] [CrossRef]
- Dreborg, S.; Sundstrom, G.; Larsson, T.; Larhammar, D. Evolution of vertebrate opioid receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 15487–15492. [Google Scholar] [CrossRef] [Green Version]
- Stevens, C. The evolution of vertebrate opioid receptors. Front. Biosci. J. Virtual Libr. 2009, 14, 1247–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundstrom, G.; Dreborg, S.; Larhammar, D. Concomitant Duplications of Opioid Peptide and Receptor Genes before the Origin of Jawed Vertebrates. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Naz, R.; Tahir, S.; Abbasi, A. An insight into the evolutionary history of human MHC paralogon. Mol. Phylogenet. Evol. 2017, 110, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A. Phylogenies of developmentally important proteins do not support the hypothesis of two rounds of genome duplication early in vertebrate history. J. Mol. Evol. 1999, 48, 565–576. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Paterson, A. MCScanX-transposed: Detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics 2013, 29, 1458–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Hagelsieb, G.; Trevino, V.; Pérez-Rueda, E.; Collado-Vides, T.S.J. Transcription unit conservation in the three domains of life: A perspective from Escherichia coli. Trends Genet. 2001, 17, 175–177. [Google Scholar] [CrossRef]
- Fani, R.; Brilli, M.; Liò, P. The Origin and Evolution of Operons: The Piecewise Building of the Proteobacterial Histidine Operon. J. Mol. Evol. 2005, 60, 378–390. [Google Scholar] [CrossRef] [PubMed]
- El-Mabrouk, N.; Noutahi, E. Bioinformatics and Phylogenetics, Seminal Contributions of Bernard Moret; Computational Biology, Chapter Gene Family Evolution: An Algorithmic Framework; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Anselmetti, Y.; Luhmann, N.; Bérard, S.; E, E.T.; Chauve, C. Comparative Genomics. In Methods in Molecular Biology; Chapter Comparative Methods for Reconstructing Ancient Genome Organization; Humana Press: New York, NY, USA, 2017; Volume 1704. [Google Scholar]
- Gagnon, Y.; Blanchette, M.; El-Mabrouk, N. A flexible ancestral genome reconstruction method based on gapped adjacencies. BMC Bioinform. 2012, 13, 1–12. [Google Scholar]
- El-Mabrouk, N.; Sankoff, D. Methods in Molecular Biology. In Evolutionary Genomics: Statistical and Computational Methods; Chapter Analysis of Gene Order Evolution Beyond Single-Copy Genes; Springer (Humana): Totowa, NJ, USA, 2012; Volume 855, pp. 397–429. [Google Scholar]
- Chauve, C.; El-Mabrouk, N.; Gueguen, L.; Semeria, M.; Tannier, E. Models and Algorithms for Genome Evolution; Computational Biology, Chapter Duplication, Rearrangement and Reconciliation: A Follow-Up 13 Years Later; Springer: London, UK, 2013. [Google Scholar]
- Renwick, J. Human Genetics; Grouchy, J., Ebling, F., Henderson, I., Eds.; Excerpta Medica: Amsterdam, The Netherlands, 1972; pp. 443–444. [Google Scholar]
- Passarge, E.; Horsthemke, B.; Farber, R. Incorrect use of the term synteny. Nat. Genet. 1999, 23, 387. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.; Heidelberg, J.; White, O.; Salzberg, S. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000, 1, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeau, J.; Taylor, B. Lengths of chromosomal segments conserved since divegence of man and mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 814–818. [Google Scholar] [CrossRef] [Green Version]
- Simison, W.B.; Parham, J.; Papenfuss, T.; Lam, A.; Henderson, J. An Annotated Chromosome-Level Reference Genome of the Red-Eared Slider Turtle. Genome Biol. Evol. 2020, 12, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Drillon, G.; Champeimont, R.; Oteri, F.; Fischer, G.; Carbone, A. Phylogenetic Reconstruction Based on Synteny Block and Gene Adjacencies. Mol. Biol. Evol. 2020, 37, 2747–2762. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Cheng, Z.; Morrison, V.; Scherer, S.; Ventura, M.; Gibbs, R.; Green, E.; Eichler, E. Recurrent duplication-driven transpositionof DNA during hominoid evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 17626–17631. [Google Scholar] [CrossRef] [Green Version]
- Mangal, M.; Srivastava, A.; Sharma, R.; Kalia, P. Conservation and Dispersion of Genes Conferring Resistance to Tomato Begomoviruses between Tomato and Pepper Genomes. Front. Plant Sci. 2017, 8, 1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, A.; Chauve, C.; Gingras, Y. Formal models of gene clusters. In Bioinformatics Algorithms: Techniques and Applications; Mandoiu, I., Zelikovsky, A., Eds.; Wiley: Hoboken, NJ, USA, 2008; Chapter 8. [Google Scholar]
- Bergeron, A.; Stoye, J. On the similarity of sets of permutations and its applications to genome comparison. J. Comput. Biol. 2003, 13, 1340–1354. [Google Scholar] [CrossRef] [Green Version]
- Heber, S.; Stoye, J. Finding all common intervals of k permutations. Lecture Notes in Computer Science. In Combinatorial Pattern Matching; Amir, A., Landau, G.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; Volume 2089, pp. 207–218. [Google Scholar]
- Uno, T.; Yagiura, M. Fast algorithms to enumerate all common intervals of two permutations. Algorithmica 2000, 26, 290–309. [Google Scholar] [CrossRef]
- Landau, G.; Parida, L.; Weimann, O. Gene proximity analysis across whole genomes via PQ trees. J. Comput. Biol. 2005, 12, 1289–1306. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, A.; Corteel, S.; Raffinot, M. The algorithmic of gene teams. In Workshop on Algorithms in Bioinformatics; Lecture Notes in Computer Science; Guigó, R., Gusfield, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 2452, pp. 464–476. [Google Scholar]
- Yang, Z.; Sankoff, D. Natural Parameter Values for Generalized Gene Adjacency. J. Comput. Biol. 2010, 17, 1113–1128. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Adam, Z.; Choi, V.; Sankoff, D. Generalized Gene Adjacencies, Graph Bandwidth, and Clusters in Yeast Evolution. IEEE/ACM Trans. Comput. Biol. Bioinform. 2009, 6, 213–220. [Google Scholar]
- Pevzner, P.; Tesler, G. Genome rearrangements in mammalian evolution: Lessons from human and mouse genomic sequences. Genome Res. 2003, 13, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Pham, S.; Pevzner, P. DRIMM-Synteny: Decomposing genomes into evolutionary conserved segments. Bioinformatics 2010, 26, 2509–2516. [Google Scholar] [CrossRef] [Green Version]
- Bader, D.; Moret, B.; Yan, M. A Linear-Time Algorithm for Computing Inversion Distance between Signed Permutations with an Experimental Study. J. Comput. Biol. 2001, 8, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, A.; Mixtacki, J.; Stoye, J. Reversal Distance without Hurdles and Fortresses. In Combinatorial Pattern Matching; Lecture Notes in Computer Science; Sahinalp, S., Muthukrishnan, S., Dogrusoz, U., Eds.; Springer: Berlin/Heidelberg, Germnay, 2004; Volume 3109, pp. 388–399. [Google Scholar]
- Tesler, G. Efficient algorithms for multichromosomal genome rearrangements. J. Comput. Syst. Sci. 2002, 65, 587–609. [Google Scholar] [CrossRef] [Green Version]
- Feijao, P.; Meidanis, J. SCJ: A breakpoint-like distance that simplifies several rearrangement problem. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, S.; Attie, O.; Friedberg, R. Efficient sorting of genomic permutations by translocation, inversion and block interchange. Bioinformatics 2005, 21, 3340–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, L.; Fertin, G.; Rusu, I. Sorting by transpositions is difficult. SIAM J. Discret. Math. 2012, 26, 1148–1180. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.; Kowada, L.; Rocco, N.; Walter, M. An Algebraic 1.375-Approximation Algorithm for the Transposition Distance Problem; Elsevier: Amsterdam, The Netherlands, 2021; Submitted. [Google Scholar]
- Sankoff, D. Minimal mutation trees of sequences. SIAM J. Appl. Math. 1975, 28. [Google Scholar] [CrossRef] [Green Version]
- Pe’er, I.; Shamir, R. The median problems for breakpoints are NP-complete. BMC Bioinform. 1998, 5. Available online: https://www.researchgate.net/profile/Ron-Shamir/publication/220138763_The_median_problems_for_breakpoints_are_NP-complete/links/02bfe50e41b4bbed55000000/The-median-problems-for-breakpoints-are-NP-complete.pdf (accessed on 8 May 2021).
- Hannenhalli, S.; Pevzner, P.A. Transforming cabbage into turnip (polynomial algorithm for sorting signed permutations by reversals). J. ACM 1999, 48, 1–27. [Google Scholar] [CrossRef]
- Bourque, G.; Pevzner, P. Genome-Scale Evolution: Reconstructing Gene Orders in the Ancestral Species. Genome Res. 2002, 12, 26–36. [Google Scholar]
- Alekseyev, M.; Pevzner, P. Breakpoint graphs and ancestral genome reconstructions. Genome Res. 2009, 19, 943–957. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Moret, B. GASTS: Parsimony Scoring under Rearrangements. In Algorithms in Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 351–363. [Google Scholar]
- Zheng, C.; Sankoff, D. On the PATHGROUPS approach to rapid small phylogeny. BMC Bioinform. 2011, 12, S4. [Google Scholar] [CrossRef] [Green Version]
- Kovac, J.; Brejova, B.; Vinar, T. A practical algorithm for ancestral rearrangement reconstruction. In International Workshop on Algorithms in Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 163–174. [Google Scholar]
- Shao, M.; Lin, Y. Approximating the edit distance for genomes with duplicate genes under DCJ, insertion and deletion. BMC Bioinform. 2012, 13, S13. [Google Scholar] [CrossRef] [Green Version]
- Willing, E.; Zaccaria, S.; Braga, M.; Stoye, J. On the inversion-indel distance. BMC Bioinform. 2013, 14, S3. [Google Scholar] [CrossRef] [Green Version]
- Braga, M.; Willing, E.; Stoye, J. Double cut and join with insertions and deletions. J. Comput. Biol. 2011, 18, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Compeau, P. DCJ-indel sorting revisited. Algorithms Mol. Biol. 2013, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Avdeyev, P.; Jiang, S.; Aganezov, S.; Hu, F.; Alekseyev, M. Reconstruction of Ancestral Genomes in Presence of Gene Gain and Loss. J. Comput. Biol. 2016, 23, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Lyubetsky, V.; Gershgorin, R.; Gorbunov, K. Chromosome structures: Reduction of certain problems with unequal gene content and gene paralogs to integer linear programming. BMC Bioinform. 2017, 18, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Fertin, G.; Labarre, A.; Rusu, I.; Tannier, E.; Vialette, S. Combinatorics of Genome Rearrangements; Istrail, S., Pevzner, P., Waterman, M., Eds.; The MIT Press: Cambridge, MA, USA; London, UK, 2009. [Google Scholar]
- Bryant, D. Comparative Genomics; Chapter the Complexity of Calculating Exemplar Distances; Kluwer Academic: Dordrecht, The Netherlands, 2000; pp. 207–211. [Google Scholar]
- Bulteau, L.; Jiang, M. Inapproximability of (1,2)-exemplar distance. In IEEE/ACM Transactions on Computational Biology and Bioinformatics; Institute of Electrical and Electronics Engineers and Association for Computing Machinery: New York City, NY, USA, 2013; pp. 1384–1390. [Google Scholar]
- Sankoff, D. Genome rearrangement with gene families. Bioinformatics 1999, 15, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Tang, J.; Schaeffer, S.; Bader, D. Exemplar or matching: Modeling DCJ problems with unequal content genome data. J. Comb. Optim. 2016, 32, 1165–1181. [Google Scholar] [CrossRef] [Green Version]
- Lorespi, D.; Tomkins, A. Block edit models for approximate string matching. Theor. Comput. Sci. 1997, 181, 159–179. [Google Scholar]
- Dondi, R.; El-Mabrouk, N. Aligning and Labeling Genomes Under the Duplication-Loss Model. In Computability in Europe (CiE); Lecture Notes in Computer Science; IOS Press: Amsterdam, The Netherlands, 2013; Volume 7921, pp. 97–107. [Google Scholar]
- Holloway, P.; Swenson, K.; Ardell, D.; El-Mabrouk, N. Ancestral Genome Organization: An Alignment Approach. J. Comput. Biol. 2013, 20, 280–295. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, S.; Reinert, K.; S, S.C. The Duplication-Loss Small Phylogeny Problem: From Cherries to Trees. J. Comput. Biol. 2013, 20, 643–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzaid, B.; Dondi, R.; El-Mabrouk, N. Duplication-Loss Genome Alignment: Complexity and Algorithm. In Proceedings of the 13th International Conference, LATA 2019, Petersburg, Russia, 26–29 March 2019. [Google Scholar]
- Bohnenkamper, L.; Braga, M.; Doerr, D.; Stoye, J. Computing the Rearrangement Distance of Natural Genomes. J. Comput. Biol. 2021, 28, 1–22. [Google Scholar] [CrossRef]
- Shao, M.; Lin, Y.; Moret, B. An exact algorithm to compute the double-cut-and-join distance for genomes with duplicate genes. J. Comput. Biol. 2015, 22, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubert, D.; Feijao, P.; Braga, M.; Stoye, J.; Martinez, F.V. Approximating the DCJ distance of balanced genomes in linear time. Algorithms Mol. Biol. 2017, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Ratan, A.; Raney, B.; Suh, B.; Miller, W.; D, D.H. The infinite sites model of genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 14254–14261. [Google Scholar] [CrossRef] [Green Version]
- Paten, B.; Zerbino, D.; Hickey, G.; Haussler, D. A unifying model of genome evolution under parsimony. BMC Bioinform. 2014, 15, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analysis with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J. MrBayes3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, F.; Patricio, M.; Muffato, M.; Pignatelli, M.; Bateman, A. TreeFam v9: A new website, more species and orthology-on-the-fly. Nucleic Acids Res. 2013. [Google Scholar] [CrossRef] [Green Version]
- Boussau, B.; Szöllősi, G.; Duret, L.; Gouy, M.; Tannier, E.; Daubin, V. Genome-scale coestimation of species and gene trees. Genome Res. 2013, 23, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szöllősi, G.J.; Rosikiewicz, W.; Boussau, B.; Tannier, E.; Daubin, V. Efficient exploration of the space of reconciled gene trees. Syst. Biol. 2013, 62, 901–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilella, A.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara gene trees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009, 19, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Capella-Gutierrez, S.; Pryszcz, L.; Denisov, I.; Kormes, D.; Marcet-Houben, M.; Gabald’on, T. PhylomeDB v3.0: An expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 2011, 39, D556–D560. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Thomas, P. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2012, 41, D377–D386. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Durand, D.; Farach-Colton, M. Notung: Dating Gene Duplications using Gene Family Trees. J. Comput. Biol. 2000, 7, 429–447. [Google Scholar] [CrossRef]
- Zhang, L. On a Mirkin-Muchnik-Smith conjecture for comparing molecular phylogenies. J. Comput. Biol. 1997, 4, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Zmasek, C.M.; Eddy, S.R. A simple algorithm to infer gene duplication and speciiation events on a gene tree. Bioinformatics 2001, 17, 821–828. [Google Scholar] [CrossRef]
- Bérard, S.; Gallien, C.; Boussau, B.; Szollosi, G.; Daubin, V.; Tannier, E. Evolution of gene neighborhoods within reconciled phylogenies. Bioinformatics 2012, 28, 382–388. [Google Scholar] [CrossRef] [Green Version]
- Noutahi, E.; Semeria, M.; Lafond, M.; Seguin, J.; Boussau, B.; Gueguen, L.; El-Mabrouk, N.; Tannier, E. Efficient Gene Tree Correction Guided by Genome Evolution. PLoS ONE 2016, 11, e0159559. [Google Scholar] [CrossRef] [Green Version]
- Patterson, M.; Szollosi, G.; Daubin, V.; Tannier, E. Lateral gene transfer, rearrangement, reconciliation. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselmetti, Y.; Berry, V.; Chauve, C.; Chateau, A.; Tannier, E.; Bérard, S. Ancestral gene synteny reconstruction improves extant species scaffolding. BMC Genom. 2015, 16, S11. [Google Scholar] [CrossRef] [Green Version]
- W, W.D.; Anselmetti, Y.; Patterson, M.; Ponty, Y.; Berard, S.; Chauve, C.; Scornavacca, C.; Daubin, V.; Tannier, E. DeCoSTAR: Reconstructing the ancestral organization of genes or genomes using reconciled phylogenies. Genome Biol. Evol. 2017, 9, 1312–1319. [Google Scholar]
- Duchemin, W. Phylogeny of Dependencies and Dependencies of Phylogenies in Genes and Genomes. Ph.D. Thesis, Université de Lyon, Lyon, France, 2017. [Google Scholar]
- Delabre, M.; El-Mabrouk, N.; Huber, K.; Lafond, M.; Mouton, V.; Noutahi, E.; Castellanos, M. Reconstructing the History of Syntenies Through Super-Reconciliation. In RECOMB-CG; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2018; pp. 179–195. [Google Scholar]
- Delabre, M.; El-Mabrouk, N.; Huber, K.; Lafond, M.; Mouton, V.; Noutahi, E.; Castellanos, M. Evolution through segmental duplications and losses: A super-Reconciliation approach. Algorithms Mol. Biol. 2020, 15, 499–506. [Google Scholar] [CrossRef]
- Aho, A.; Yehoshua, S.; Szymanski, T.; Ullman, J. Inferring a tree from lowest common ancestors with an application to the optimization of relational expressions. SIAM J. Comput. 1981, 10, 405–421. [Google Scholar] [CrossRef]
- Constantinescu, M.; Sankoff, D. An efficient algorithm for supertrees. J. Classif. 1995, 12, 101–112. [Google Scholar] [CrossRef]
- Ng, M.; Wormald, N. Reconstruction of rooted trees from subtrees. Discret. Appl. Math. 1996, 69, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Semple, C. Reconstructing minimal rooted trees. Discret. Appl. Math. 2003, 127, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Bryant, D. A classification of consensus methods for phylogenetics. DIMACS Ser. Discret. Math. Theor. Comput. Sci. 2003, 61, 163–184. [Google Scholar]
- Lafond, M.; El-Mabrouk, N.; Huber, K.; Moulton, V. The complexity of comparing multiply-labelled trees by extending phylogenetic-tree metrics. Theor. Comput. Sci. 2019, 760, 15–34. [Google Scholar] [CrossRef] [Green Version]
- Jansson, J.; Lemence, R.; Lingas, A. The complexity of inferring a minimally resolved phylogenetic supertree. SIAM J. Comput. 2012, 41, 272–291. [Google Scholar] [CrossRef] [Green Version]
- Huber, K.; Moulton, V.; Spillner, A. Computing a consensus of multilabeled trees. In Proceedings of the 14th Workshop on Algorithm Engineering and Experiments (ALENEX 2012), Kyoto, Japan, 16 January 2012; pp. 84–92. [Google Scholar]
- Lott, M.; Spillner, A.; Huber, K. Inferring polyploid phylogenies from multiply-labeled gene trees. BMC Evol. Biol. 2009, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Gascon, M.; Dondi, R.; El-Mabrouk, N. Complexity and algorithm for MUL-tree pruning. In Proceedings of the IWOCA 2021—32nd International Workshop on Combinatorial Algorithms, Ottawa, ON, Canada, 5–7 July 2021. Lecture Notes in Computer Science. [Google Scholar]
- Paszek, J.; Gorecki, P. Efficient Algorithms for Genomic Duplication Models. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 15, 1515–1524. [Google Scholar] [CrossRef]
- Fellows, M.; Hallet, M.; Stege, U. On the multiple gene duplication problem. In Proceedings of the 9th International Symposium on Algorithms and Computation, Taejon, Korea, 14–16 December 1998; pp. 347–356. [Google Scholar]
- Paszek, J.; Górecki, P. Genomic duplication problems for unrooted gene trees. BMC Genom. 2016, 17, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Guigo, R.; Muchnik, I.; Smith, T. Reconstruction of Ancient Molecular Phylogeny. Mol. Phylogenet. Evol. 1996, 6, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Czabarka, E.; Szkély, L.; Vision, T. Minimizing the number of episodes and Gallai’s theorem on intervals. arXiv 2012, arXiv:1209.5699. [Google Scholar]
- Page, R.; Cotton, J. Vertebrate Phylogenomics: Reconciled Trees and Gene Duplications. Pac. Symp. Biocomput. 2002, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Bansal, M.S.; Eulenstein, O. The multiple gene duplicationproblem revisited. Bioinformatics 2008, 24, i132–i138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burleigh, J.G.; Bansal, M.S.; Wehe, A.; Eulenstein, O. Locating multiple gene duplications through reconciled trees. In Proceedings of the Research in Computational Molecular Biology, Singapore, 30 March–2 April 2008; Volume 4955, pp. 273–284. [Google Scholar]
- Luo, C.; Chen, M.; Chen, Y.; Yang, R.W.L.; Liu, H.; Chao, K. Linear-time algorithms for the multiple gene duplication problems. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 260–265. [Google Scholar]
- Paszek, J.; Górecki, P. Inferring duplication episodes from unrooted gene trees. BMC Genom. 2018, 19, 288. [Google Scholar] [CrossRef] [Green Version]
- Dondi, R.; Lafond, M.; Scornavacca, C. Reconciling Multiple Genes Trees via Segmental Duplications and Losses. Algorithms Mol. Biol. 2019, 14, 1–9. [Google Scholar] [CrossRef]
- Zhou, L.; Huang, B.; Meng, X.; Wang, G.; Wang, F.; Xu, Z.; Song, R. The amplification and evolution of orthologous 22-kDa α-prolamin tandemly arrayed genes in coix, sorghum and maize genomes. Plant Mol. Biol. 2010, 74, 631–643. [Google Scholar] [CrossRef]
- Shoja, V.; Zhang, L. A Roadmap of Tandemly Arrayed Genes in the Genomes of Human, Mouse, and Rat. Mol. Biol. Evol. 2006, 23, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zhang, L.; Vinar, T.; Miller, W. Inferring the recent duplication history of a gene cluster. In Comparative Genomics; Lecture Notes in Computer Science; Ciccarelli, F., Miklós, I., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 5817. [Google Scholar]
- Vinař, T.; Brejová, B.; Song, G.; Siepel, A. Reconstructing Histories of Complex Gene Clusters on a Phylogeny. J. Comput. Biol. 2010, 17, 1267–1269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Song, G.; Hsu, C.; Miller, W. Simultaneous History Reconstruction for Complex Gene Clusters in Multiple Species. Available online: https://www.worldscientific.com/doi/abs/10.1142/9789812836939_0016 (accessed on 8 April 2021).
- Fitch, W. Phylogenies constrained by cross-over process as illustrated by human hemoglobins and a thirteen-cycle, eleven amino-acid repeat in human apolipoprotein A-I. Genetics 1977, 86, 623–644. [Google Scholar] [CrossRef]
- Bertrand, D.; Gascuel, O. Topological rearrangements and local search method for tandem duplication trees. IEEE/ACM Trans. Comput. Biol. Bioinform. 2005, 2, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, D.; Lajoie, M.; El-Mabrouk, N. Inferring Ancestral Gene Orders for a Family of Tandemly Arrayed Genes. J. Comput. Biol. 2008, 15, 1063–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Ratan, A.; Raney, B.J.; Sush, B.B.; Zhang, L.; Miller, W.; Haussler, D. DUPCAR: Reconstructing Contiguous Ancestral Regions with Duplication. J. Comput. Biol. 2008, 15, 1007–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajoie, M.; Bertrand, D.; El-Mabrouk, N. Inferring the Evolutionary History of Gene Clusters from Phylogenetic and Gene Order Data. Mol. Biol. Evol. 2010, 27, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Savard, O.T.; Bertrand, D.; El-Mabrouk, N. Evolution of orthologous tandemly arrayed gene clusters. BMC Bioinform. 2011, 12, S2. [Google Scholar]
- Doyon, J.P.; Scornavacca, C.; Gorbunov, K.Y.; Szöllősi, G.J.; Ranwez, V.; Berry, V. An efficient algo. for gene/species trees parsimonious reconciliation with losses, duplications and transfers. In RECOMB-CG; Lecture Notes in Computer Science; Springer: Berlin, Germany, 2010; Volume 6398, pp. 93–108. [Google Scholar]
- Doyon, J.; Ranwez, V.; Daubin, V.; Berry, V. Models, algorithms and programs for phylogeny reconciliation. Briefings Bioinform. 2011, 12, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallett, M.; Lagergren, J. Efficient algorithms for lateral gene transfer problems. In Proceedings of the the Fifth Annual International Conference on Computational Biology, Montreal, QC, Canada, 22–25 April 2001; pp. 149–156. [Google Scholar]
- Ovadia, Y.; Fielder, D.; Conow, C.; Libeskind-Hadas, R. The cophylogeny reconstruction problem is NP-complete. J. Comput. Biol. 2011, 18, 59–65. [Google Scholar] [CrossRef]
- Bansal, M.; Alm, E.; Kellis, M. Efficient algorithms for the reconciliation problem with gene duplication, horizontal transfer and loss. Bioinformatics 2012, 28, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Tofigh, A.; Hallett, M.; Lagergren, J. Simultaneous identification of duplications and lateral gene transfers. IEEE/ACM Trans. Comput. Bioinform. 2011, 8, 517–535. [Google Scholar] [CrossRef]
- Tofigh, A. Using Trees to Capture Reticulate Evolution: Lateral Gene Transfers and Cancer Progression. Ph.D. Thesis, KTH Royal Institute of Technology, Stockholm, Sweden, 2009. [Google Scholar]
- David, L.; Alm, E. Rapid evolutionary innovation during an Archaean genetic expansion. Nature 2011, 469, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libeskind-Hadas, R.; Charleston, M. On the computational complexity of the reticulate cophylogeny reconstruction problem. J. Comput. Biol. 2009, 16, 105–117. [Google Scholar] [CrossRef]
- Anselmetti, Y.; El-Mabrouk, N.; Lafond, M.; Ouandraoua, A. Gene Tree and Species Tree Reconciliation with Endosymbiotic Gene Transfer. Available online: http://www-labs.iro.umontreal.ca/~mabrouk/Publications/ISMB2021.pdf (accessed on 8 April 2021).
- Bollback, J. SIMMAP: Stochastic character mapping of discrete traits onphylogenies. BMC Bioinform. 2006, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.; Nielsen, R.; Bollback, J. Stochastic mapping of morphological characters. Syst. Biol. 2003, 52, 131–158. [Google Scholar] [CrossRef]
- Simon, D.; Larget, B. Bayesian Analysis to Describe Genomic Evolution by Rearrangement (BADGER); Version 1.02 Beta; Department of Mathematics and Computer Science, Duquesne University: Pittsburgh, PA, USA, 2004. [Google Scholar]
- Roger, A.; Munoz-Gomez, S.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.B.; Warren, J.M.; Williams, A.M.; Wu, Z.; Abdel-Ghany, S.E.; Chicco, A.J.; Havird, J.C. Cytonuclear integration and co-evolution. Nat. Rev. Genet. 2018, 19, 635–648. [Google Scholar] [CrossRef]
- Brandvain, Y.; Wade, M. The Functional Transfer of Genes From the Mitochondria to the Nucleus: The Effects of Selection, Mutation, Population Sizeand Rate of Self-Fertilization. Genetics 2009, 182, 1129–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S. The economics of endosymbiotic gene transfer and the evolution of organellar genomes. bioRxiv 2020. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Mabrouk, N. Predicting the Evolution of Syntenies—An Algorithmic Review. Algorithms 2021, 14, 152. https://doi.org/10.3390/a14050152
El-Mabrouk N. Predicting the Evolution of Syntenies—An Algorithmic Review. Algorithms. 2021; 14(5):152. https://doi.org/10.3390/a14050152
Chicago/Turabian StyleEl-Mabrouk, Nadia. 2021. "Predicting the Evolution of Syntenies—An Algorithmic Review" Algorithms 14, no. 5: 152. https://doi.org/10.3390/a14050152
APA StyleEl-Mabrouk, N. (2021). Predicting the Evolution of Syntenies—An Algorithmic Review. Algorithms, 14(5), 152. https://doi.org/10.3390/a14050152