Complete Chloroplast Genome of Fokienia hodginsii (Dunn) Henry et Thomas: Insights into Repeat Regions Variation and Phylogenetic Relationships in Cupressophyta

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Genome Assembly
2.2. Genome Annotation and Sequence Analyses
2.3. Repeat Sequence Statistics and IR Identification
2.4. Phylogenetic Research
3. Results
3.1. Genome Features
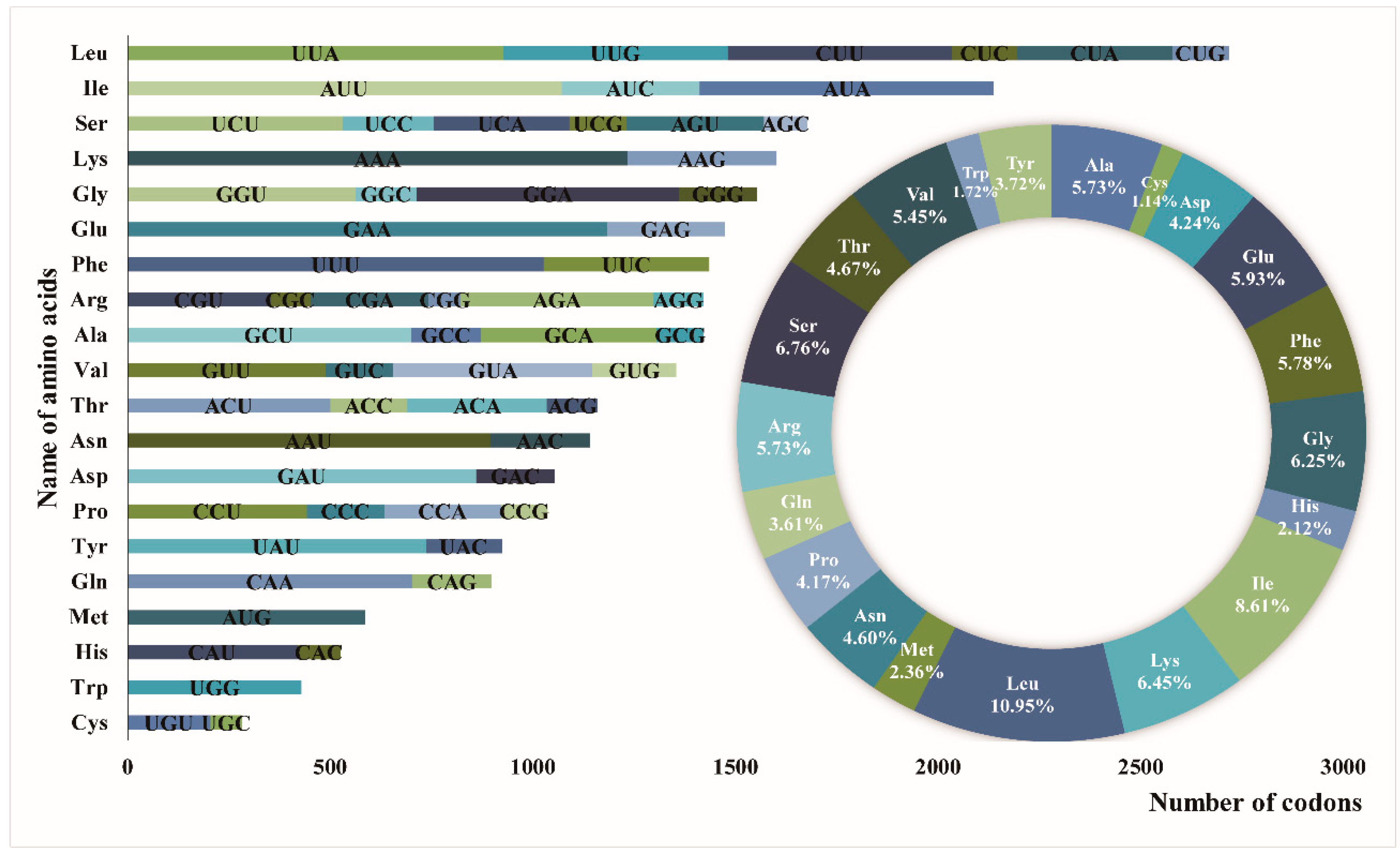
3.2. Repeat Sequence and SSR Analysis
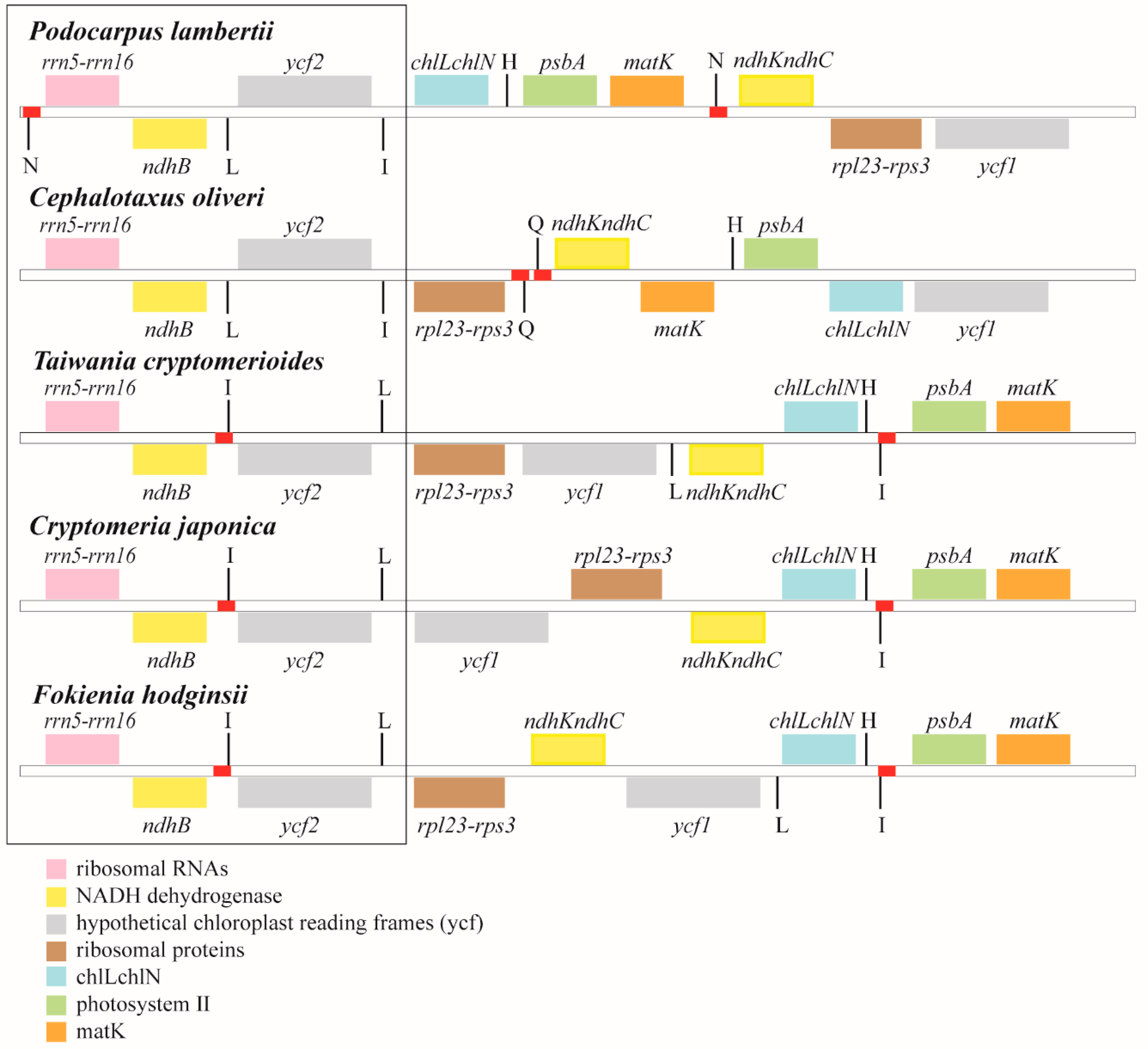
3.3. Residual Inverted Repeat (IR) Regions
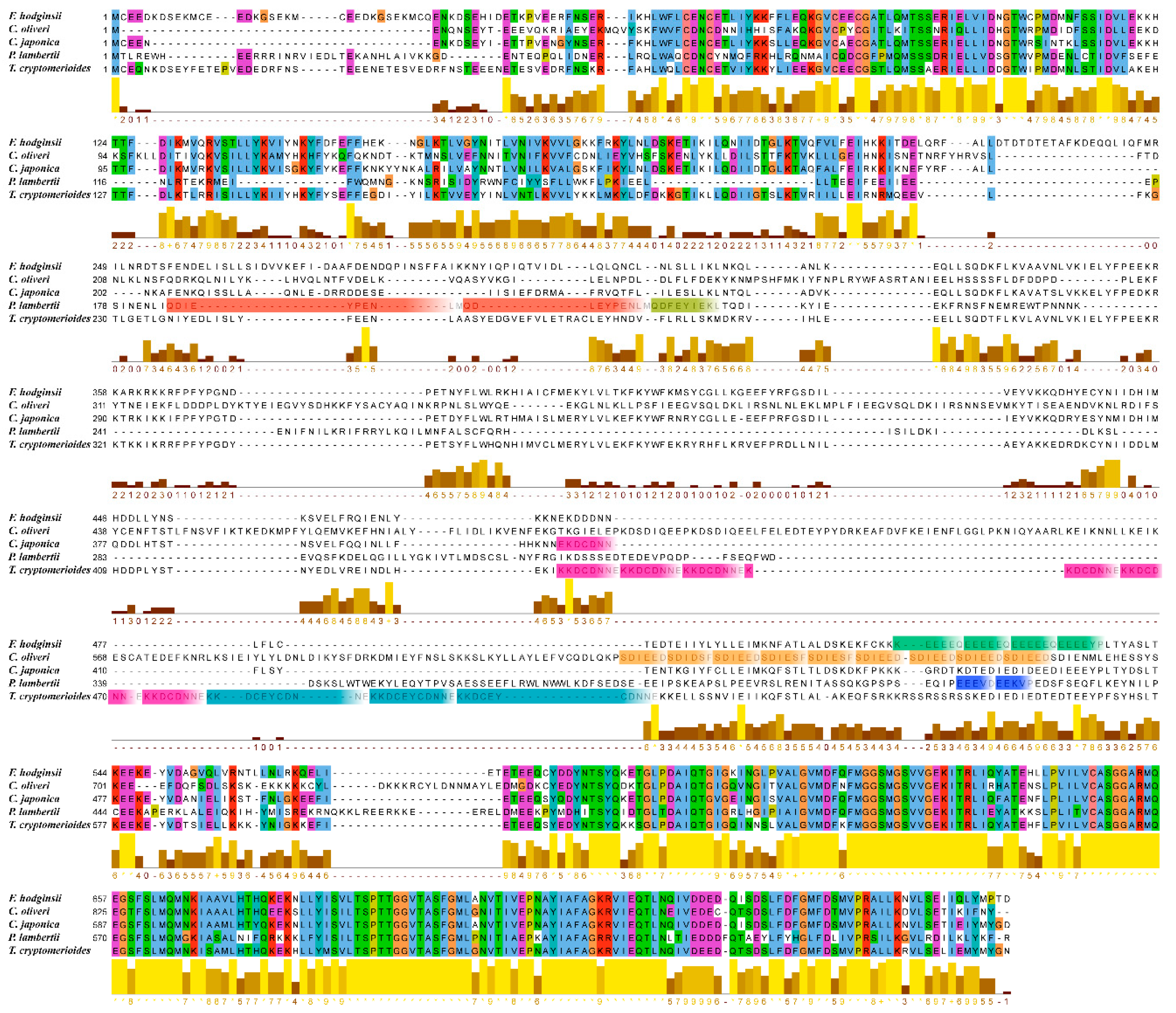
3.4. Phylogenetic Research
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, Z.W. A precious timber species—Fokienia hodginsii. J. Fujian For. Sci. Tech. 1994, 21, 62–66. [Google Scholar]
- Hou, B.X.; YU, G.F.; Lin, F.; Cheng, Z.H. Study on Fokienia hodginsii natural wild wood community. Hunan For. Sci. Tech. 2004, 31. [Google Scholar]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Shinozaki, K.Y.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. Plant Mol. Biol. Rep. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Ohyama, K.; Fukuzawa, H.; Kohchi, T.; Shirai, H.; Sano, T.; Sano, S.; Umesono, K.; Shiki, Y.; Takeuchi, M.; Chang, Z.; et al. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 1986, 322, 572–574. [Google Scholar] [CrossRef]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: Diversified genomic structure of coniferous species. BMC Plant Biol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Comparative Organization of Chloroplast Genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef]
- Sugiura, M. The chloroplast chromosomes in land plants. Annu. Rev. Cell Biol. 1989, 5, 51–70. [Google Scholar] [CrossRef]
- Jansen, R.K.; Ruhlman, T.A. Plastid Genomes of Seed Plants. In Genomics of Chloroplasts and Mitochondria; Springer: Dordrecht, The Netherlands, 2012; Volume 35, pp. 103–126. [Google Scholar]
- Strauss, S.H.; Palmer, J.D.; Howe, G.T.; Doerksen, A.H. Chloroplast genomes of two conifers lack a large inverted repeat and are extensively rearranged. Proc. Natl. Acad. Sci. USA 1988, 85, 3898–3902. [Google Scholar] [CrossRef]
- Palmer, J.D.; Thompson, W.F. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 1982, 29, 537–550. [Google Scholar] [CrossRef]
- Palmer, J.D.; Stein, D.B. Conservation of chloroplast genome structure among vascular plants. Curr. Genet. 1986, 10, 823–833. [Google Scholar] [CrossRef]
- Lin, C.P.; Wu, C.S.; Huang, Y.Y.; Chaw, S.M. The Complete Chloroplast Genome of Ginkgo biloba Reveals the Mechanism of Inverted Repeat Contraction. Genome Biol. Evol. 2012, 4, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Drescher, A.; Ruf, S.; Calsa, J.T.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2010, 22, 97–104. [Google Scholar] [CrossRef]
- Wu, C.S.; Wang, Y.N.; Hsu, C.Y.; Lin, C.P.; Chaw, S.M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Chloroplast DNA Evolution and Biosystematic Uses of Chloroplast DNA Variation. Am. Nat. 1987, 130, 6–29. [Google Scholar] [CrossRef]
- Sandbrink, J.; Vellekoop, P.; Van, H.R.; Van, B.J. A method for evolutionary studies on RFLP of chloroplast DNA, applicable to a range of plant species. Biochem. Syst. Ecol. 1989, 17, 45–49. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2008, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 7. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerch, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2 - A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Braukmann, T.W.; Kuzmina, M.; Stefanović, S. Loss of all plastid ndh genes in Gnetales and conifers: Extent and evolutionary significance for the seed plant phylogeny. Curr. Genet. 2009, 55, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Hildebrand, M.; Hallick, R.B.; Passavant, C.W.; Bourque, D.P. Trans-splicing in chloroplasts: The rps 12 loci of Nicotiana tabacum. Proc. Natl. Acad. Sci. USA 1988, 85, 372–376. [Google Scholar] [CrossRef]
- Shimada, H.; Sugiura, M. Fine structural features of the chloroplast genome: Comparison of the sequenced chloroplast genomes. Nucleic Acids Res. 1991, 19, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Eun, H.M. Enzymes and Nucleic Acids: General Principles. In Enzymology Primer for Recombinant DNA Technology; Elsevier: Amsterdam, The Netherlands, 1996; pp. 1–108. [Google Scholar]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Cardle, L.; Ramsay, L.; Milbourne, D.; Macaulay, M.; Marshall, D.; Waugh, R. Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 2000, 156, 847–854. [Google Scholar] [PubMed]
- Madoka, Y.; Tomizawa, K.I.; Mizoi, J.; Nishida, I.; Nagano, Y.; Sasaki, Y. Chloroplast transformation with modified accD operon increases acetyl-CoA carboxylase and causes extension of leaf longevity and increase in seed yield in tobacco. Plant Cell Physiol. 2002, 43, 1518. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Su, Y.J.; Wang, T. The Repeat Sequences and Elevated Substitution Rates of the Chloroplast accD Gene in Cupressophytes. Frontiers Plant Sci. 2018, 9, 533. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, J.; Yang, B.; Li, R.; Zhu, W.; Sun, L.; Tian, J.; Zhang, L. The complete chloroplast genome sequence of Taxus chinensis var. mairei (Taxaceae): Loss of an inverted repeat region and comparative analysis with related species. Gene 2014, 540, 201–209. [Google Scholar] [CrossRef]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Frontiers Plant Sci. 2015, 6, 447. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, D.Z. Advances in Phylogenomics Based on Complete Chloroplast Genomes. Plant Diver. Resour. 2011, 33, 365–375. [Google Scholar]
- Chaw, S.M.; Parkinson, C.L.; Cheng, Y.; Vincent, T.M.; Palmer, J.D. Seed plant phylogeny inferred from all three plant genomes: Monophyly of extant gymnosperms and origin of Gnetales from conifers. Proc. Natl. Acad. Sci. USA 2000, 97, 4086–4091. [Google Scholar] [CrossRef] [Green Version]
- Conran, J.G.; Wood, G.M.; Martin, P.G.; Dowd, J.M.; Quinn, C.J.; Gadek, P.A.; Price, R.A. Generic relationships within and between the gymnosperm families Podocarpaceae and Phyllocladaceae based on an analysis of the chloroplast gene rbcL. Aust. J. Bot. 2000, 48, 715–724. [Google Scholar] [CrossRef]
- Wang, X.Q.; Shu, Y.Q. Chloroplast matK gene phylogeny of Taxaceae and Cephalotaxaceae, with additional reference to the systematic position of Nageia. Acta Phytotax. Sin. 2000, 38, 201–210. [Google Scholar]
- Setoguchi, H.; Osawa, T.A.; Pintaud, J.C.; Jaffre, T.; Veillon, J.M. Phylogenetic Relationships within Araucariaceae Based on rbcL Gene Sequences. Am. J. Bot. 1998, 85, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, P.; Wagstaff, B. The Southern Conifer Family Araucariaceae: History, Status, and Value for Paleoenvironmental Reconstruction. Annu. Rev. Ecol. Syst. 2001, 32, 397–414. [Google Scholar] [CrossRef] [Green Version]
- Chase, M.W.; Soltis, D.E.; Olmstead, R.G.; Morgan, D.; Les, D.H.; Mishler, B.D.; Duvall, M.R.; Price, R.A.; Hills, H.G.; Qiu, Y.L.; et al. Phylogenetics of Seed Plants: An Analysis of Nucleotide Sequences from the Plastid Gene rbcL. Ann. Mo. Bot. Gard. 1993, 80, 528–548. [Google Scholar] [CrossRef]
- Hsu, C.Y.; Wu, C.S.; Chaw, S.M. Ancient nuclear plastid DNA in the yew family (taxaceae). Genome Biol. Evol. 2014, 6, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.X.; Ye, Y.J.; Bai, T.; Xu, M.; Xu, L.A. Complete Chloroplast Genome of Pinus massoniana (Pinaceae): Gene Rearrangements, Loss of ndh Genes, and Short Inverted Repeats Contraction, Expansion. Molecules 2017, 22, 1528. [Google Scholar] [CrossRef] [PubMed]
- Celiński, K.; Kijak, H.; Barylski, J.; Grabsztunowicz, M.; Wojnicka-Półtorak, A.; Chudzińska, E. Characterization of the complete chloroplast genome of Pinus uliginosa (Neumann) from the Pinus mugo complex. Conserv. Genet. Resour. 2016, 9, 1–4. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Shahzad, R.; Lubna; Kang, S.M.; Harrasi, A.A.; Rawahi, A.A.; Lee, I.J. Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinus taeda L.) with related species. PLoS ONE 2018, 13, e0192966. [Google Scholar] [CrossRef]
- Vieira, L.N.; Faoro, H.; Rogalski, M.; Fraga, H.P.F.; Cardoso, R.L.A.; Souza, E.M.; Pedrosa, F.O.; Nodari, R.O.; Guerra, M.P. The complete chloroplast genome sequence of Podocarpus lambertii: Genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 2014, 9, e90618. [Google Scholar] [CrossRef]
- Yi, X.; Gao, L.; Wang, B.; Su, Y.J.; Wang, T. The Complete Chloroplast Genome Sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary Comparison of Cephalotaxus Chloroplast DNAs and Insights into the Loss of Inverted Repeat Copies in Gymnosperms. Genome Biol. Evol. 2013, 5, 688–698. [Google Scholar] [CrossRef]
- Tsudzuki, J.; Nakashima, K.; Tsudzuki, T.; Hiratsuka, J.; Shibata, M.; Wakasugi, T.; Sugiura, M. Chloroplast DNA of black pine retains a residual inverted repeat lacking rRNA genes: Nucleotide sequences of trnQ, trnK, psbA, trnI and trnH and the absence of rps16. Mol. Gen. Genet. 1992, 232, 206–214. [Google Scholar] [PubMed]
- Hart, J.A. A CLADISTIC ANALYSIS OF CONIFERS: PRELIMINARY RESULTS. J Arnold. Arbor. 1987, 68, 269–307. [Google Scholar]
- Chaw, S.M.; Zharkikh, A.; Sung, H.M.; Lau, T.C.; Li, W.H. Molecular phylogeny of extant gymnosperms and seed plant evolution: Analysis of nuclear 18S rRNA sequences. Mol. Biol. Evol. 1997, 14, 56–68. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Category | Group of Genes | Gene Names |
|---|---|---|
| Self-replication | Ribosomal RNA genes | rrn4.5, rrn5, rrn16, rrn23 |
| Transfer RNA genes | trnP-GGG, trnL-UAG, trnH-GUG, trn-I CAU(×2), trnK-UUU*, trnQ-UUG(×2), trnS-GCU, trnG-GCC, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-UCC*, trnfM-CAU, trnS-GGA, trnT-UGU, trnL-UAA*, trnF-GAA, trnN-GUU, trnR-ACG, trnA-UGC*, trnI-GAU*, trnV-GAC, trnL-CAA, trnW-CCA, trnP-UGG, trnV-UAC*, trnM-CAU | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7, rps8, rps11, rps12**, rps14, rps15, rps16*, rps18, rps19 | |
| Large subunit of ribosome | rpl2*, rpl14, rpl16*, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1*, rpoC2 | |
| Translational initiation factor | infA | |
| Genes for photosynthesis | Subunits of photosystem Ⅰ | psaA, psaB, psaC, psaI, psaJ, psaM, ycf3**, ycf4 |
| Subunits of photosystem Ⅱ | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of cytochrome | petA, petB*, petD, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF*, atpH, atpI | |
| Chlorophyll biosynthesis | chlB, chlL, chlN | |
| Large subunit of Rubisco | rbcL | |
| Subunits of NADH dehydrogenase | ndhA*, ndhB*, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Other genes | Maturase | matK |
| Envelope membrane protein | cemA | |
| Subunit of acetyl-CoA | accD | |
| C-type cytochrome synthesis gene | ccsA | |
| Protease | clpP | |
| Component of TIC complex | ycf1 | |
| Genes of unknown function | Conserved open reading frames | ycf2 |
| Gene | Exon Ⅰ (bp) | Intron Ⅰ (bp) | Exon Ⅱ (bp) | Intron Ⅱ (bp) | Exon Ⅲ (bp) |
|---|---|---|---|---|---|
| atpF | 145 | 660 | 410 | ||
| ndhA | 558 | 745 | 540 | ||
| ndhB | 711 | 659 | 756 | ||
| rpl2 | 398 | 643 | 433 | ||
| rpl16 | 9 | 824 | 411 | ||
| rps12 | 114 | / | 232 | 526 | 26 |
| rps16 | 40 | 235 | 41 | ||
| rpoC1 | 442 | 726 | 1673 | ||
| petB | 6 | 770 | 642 | ||
| ycf3 | 126 | 688 | 226 | 690 | 158 |
| trnA-UGC | 38 | 776 | 35 | ||
| trnG-UCC | 23 | 739 | 48 | ||
| trnI-GAU | 42 | 892 | 35 | ||
| trnK-UUU | 37 | 2457 | 35 | ||
| trnL-UAA | 35 | 438 | 50 | ||
| trnV-UAC | 39 | 518 | 37 |
| T(U)% | C% | A% | G% | Length (bp) | Number | GC (%) | |
|---|---|---|---|---|---|---|---|
| Total | 31.9 | 17.6 | 33.2 | 17.2 | 129,534 | - | 34.8 |
| CDS | 31.8 | 16.6 | 32.0 | 19.6 | 74,715 | - | 36.2 |
| First codon position | 24 | 18.1 | 30.8 | 27.5 | - | 24,905 | 45.6 |
| Second codon position | 33 | 19.5 | 30.8 | 16.6 | - | 24,905 | 36.1 |
| Third codon position | 39 | 12.1 | 34.3 | 14.8 | - | 24,905 | 26.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zang, M.; Su, Q.; Weng, Y.; Lu, L.; Zheng, X.; Ye, D.; Zheng, R.; Cheng, T.; Shi, J.; Chen, J. Complete Chloroplast Genome of Fokienia hodginsii (Dunn) Henry et Thomas: Insights into Repeat Regions Variation and Phylogenetic Relationships in Cupressophyta. Forests 2019, 10, 528. https://doi.org/10.3390/f10070528
Zang M, Su Q, Weng Y, Lu L, Zheng X, Ye D, Zheng R, Cheng T, Shi J, Chen J. Complete Chloroplast Genome of Fokienia hodginsii (Dunn) Henry et Thomas: Insights into Repeat Regions Variation and Phylogenetic Relationships in Cupressophyta. Forests. 2019; 10(7):528. https://doi.org/10.3390/f10070528
Chicago/Turabian StyleZang, Mingyue, Qian Su, Yuhao Weng, Lu Lu, Xueyan Zheng, Daiquan Ye, Renhua Zheng, Tielong Cheng, Jisen Shi, and Jinhui Chen. 2019. "Complete Chloroplast Genome of Fokienia hodginsii (Dunn) Henry et Thomas: Insights into Repeat Regions Variation and Phylogenetic Relationships in Cupressophyta" Forests 10, no. 7: 528. https://doi.org/10.3390/f10070528
APA StyleZang, M., Su, Q., Weng, Y., Lu, L., Zheng, X., Ye, D., Zheng, R., Cheng, T., Shi, J., & Chen, J. (2019). Complete Chloroplast Genome of Fokienia hodginsii (Dunn) Henry et Thomas: Insights into Repeat Regions Variation and Phylogenetic Relationships in Cupressophyta. Forests, 10(7), 528. https://doi.org/10.3390/f10070528