
Changes in Soil Microbial Community and Carbon Flux Regime across a Subtropical Montane Peatland-to-Forest Successional Series in Taiwan


Abstract
:1. Introduction
2. Materials and Methods
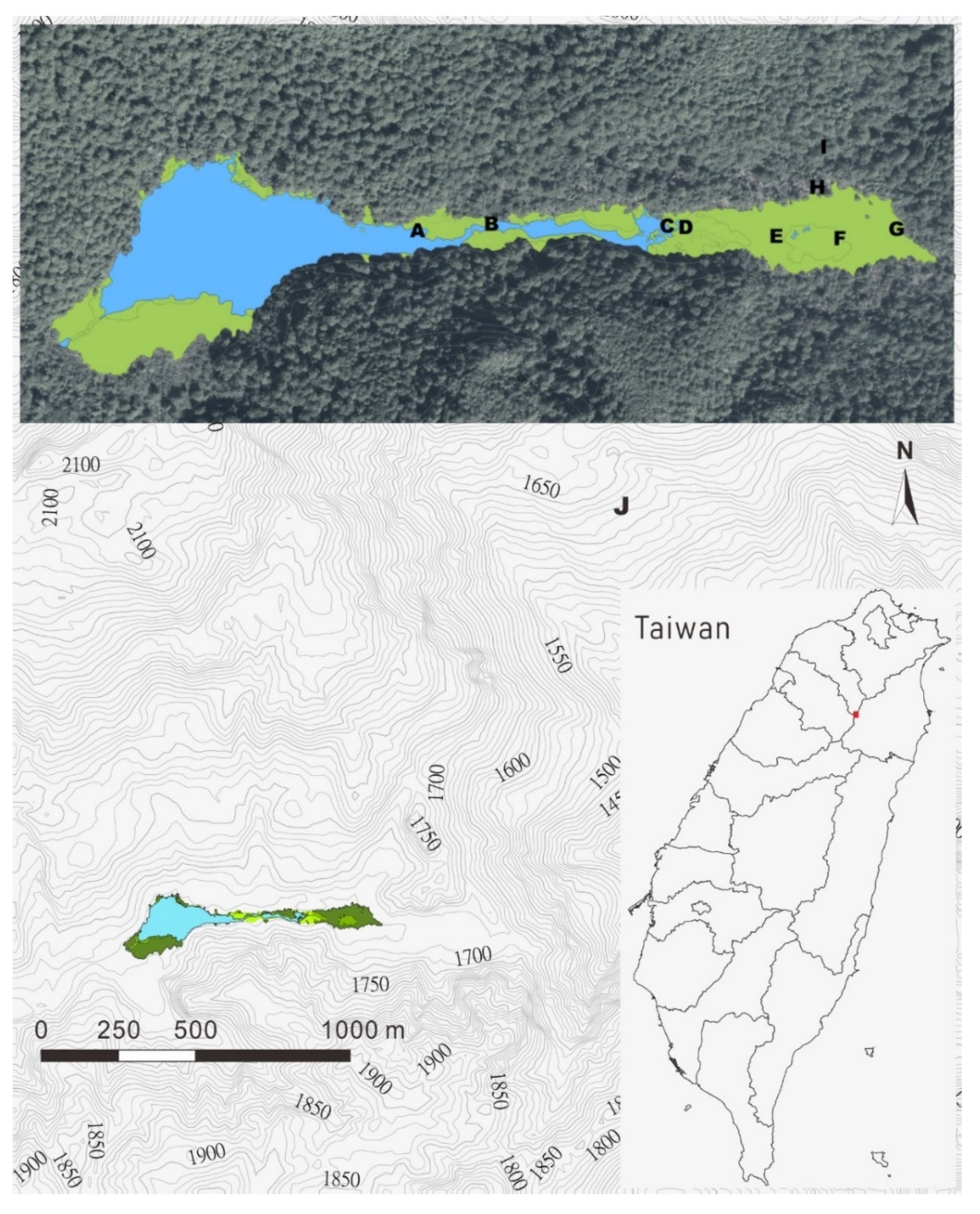
2.1. Site Description
2.2. Plant Community Survey
2.3. Microbial Characterization
2.4. Analysis of Microbial Community
2.5. Surface CO2 and CH4 Fluxes
2.5.1. Flux Measurement
2.5.2. Analysis of Carbon Flux Data
3. Results
3.1. Plant Community
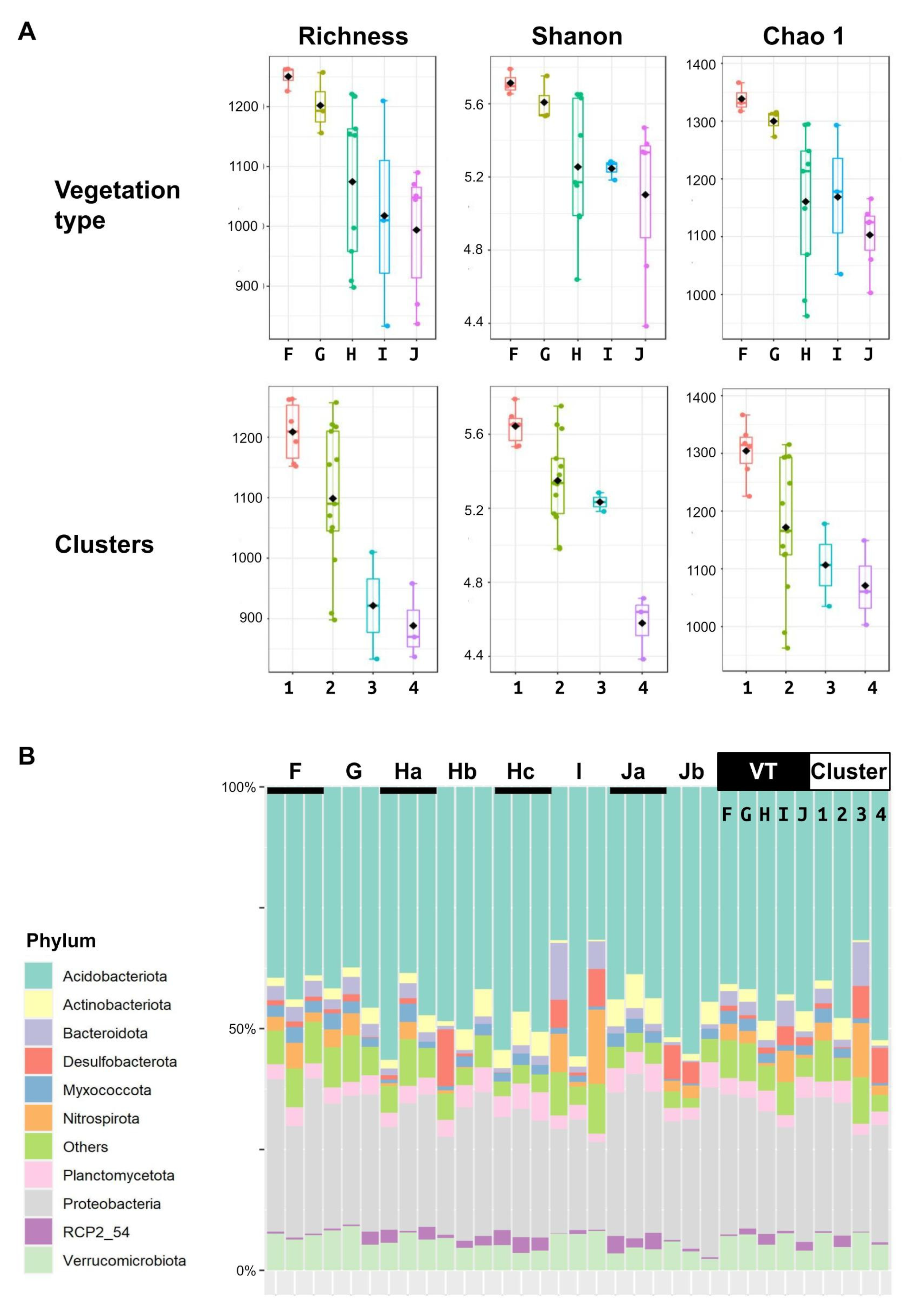
3.2. Phylogenetic Characterization of Microbiota
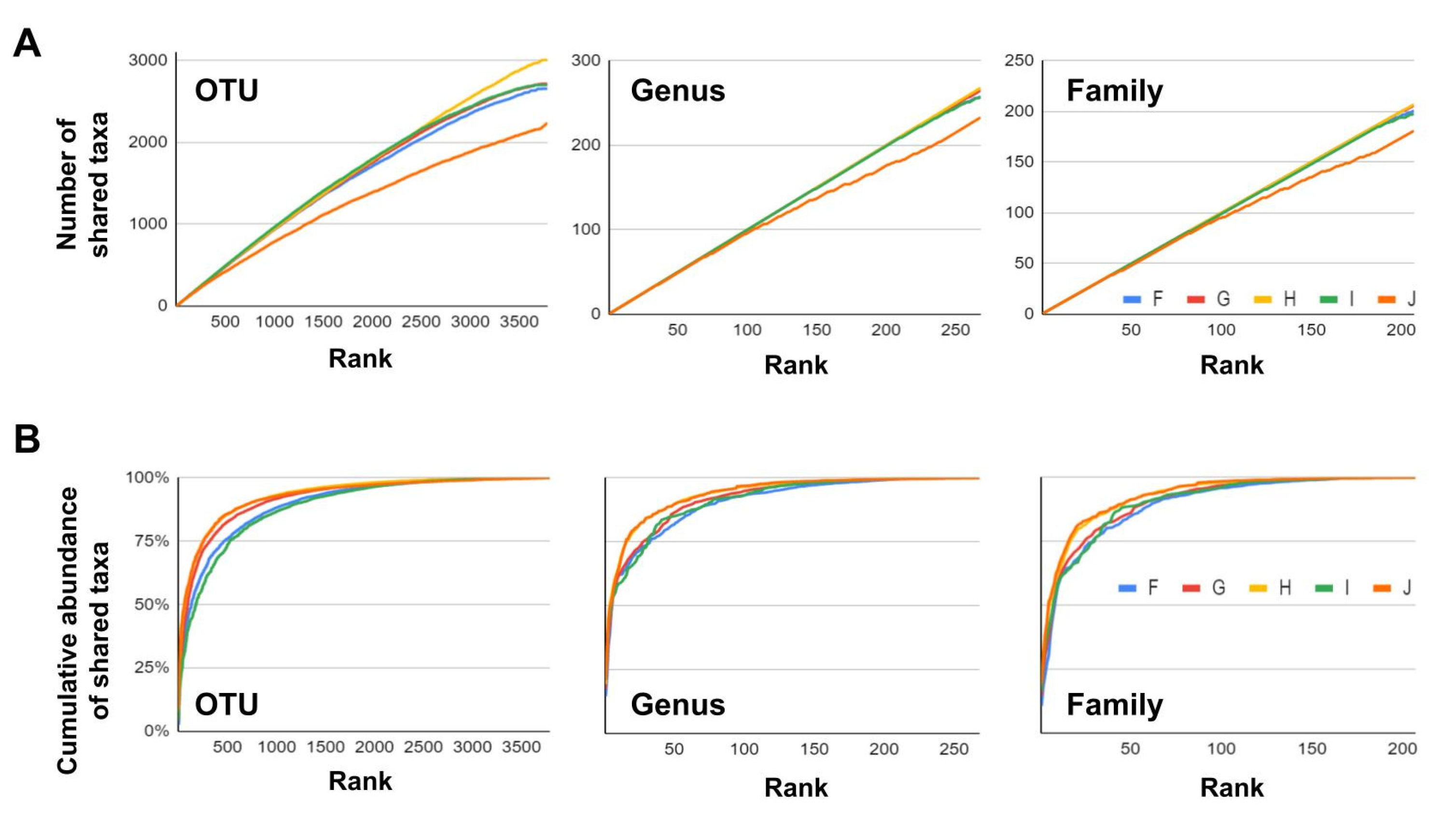
3.3. Conservation of Taxa within the YYL Watershed
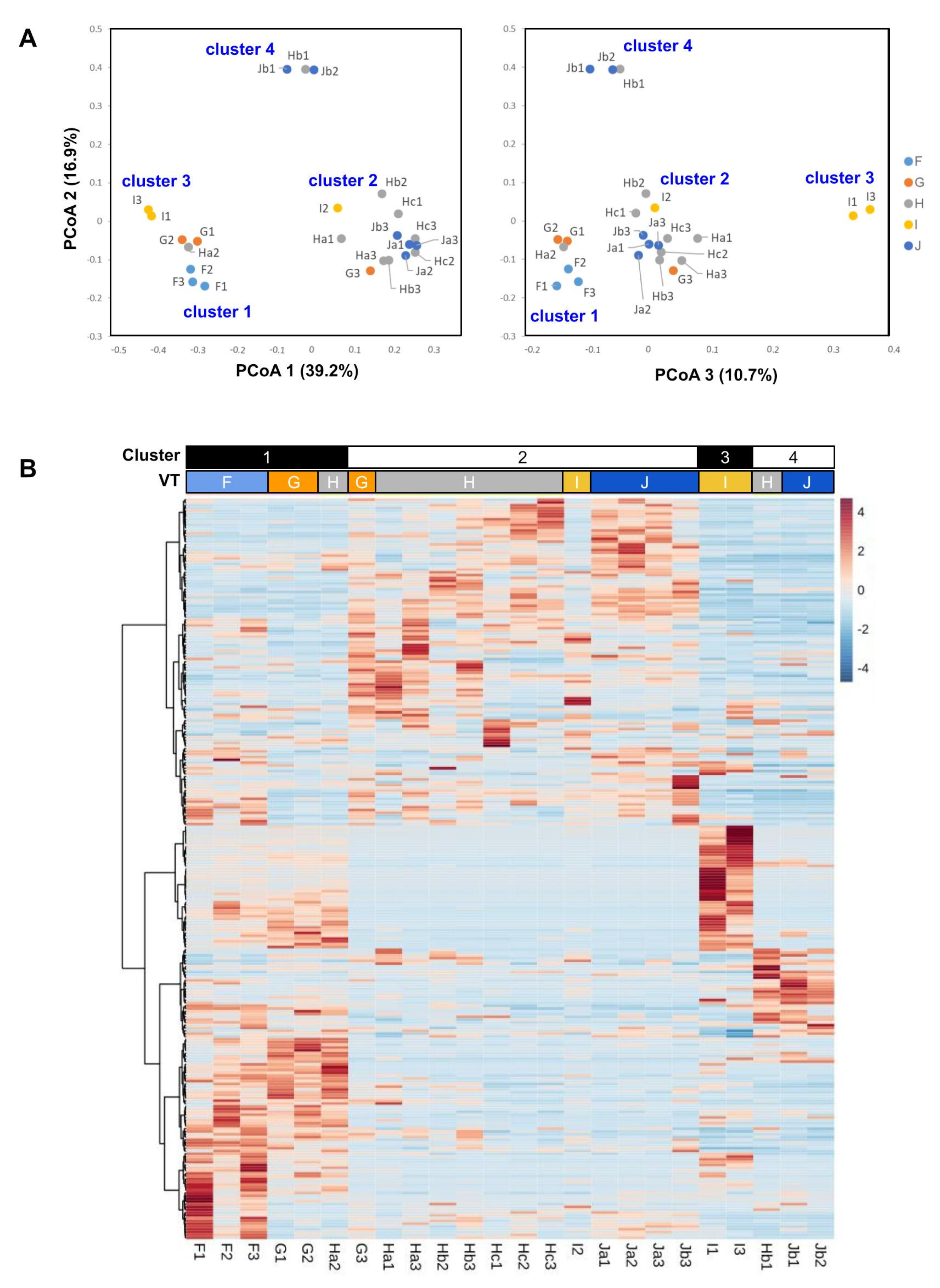
3.4. Microbial Community Types
3.5. Microbiota along the Bog-to-Forest Gradient
3.6. Methanotrophic Community
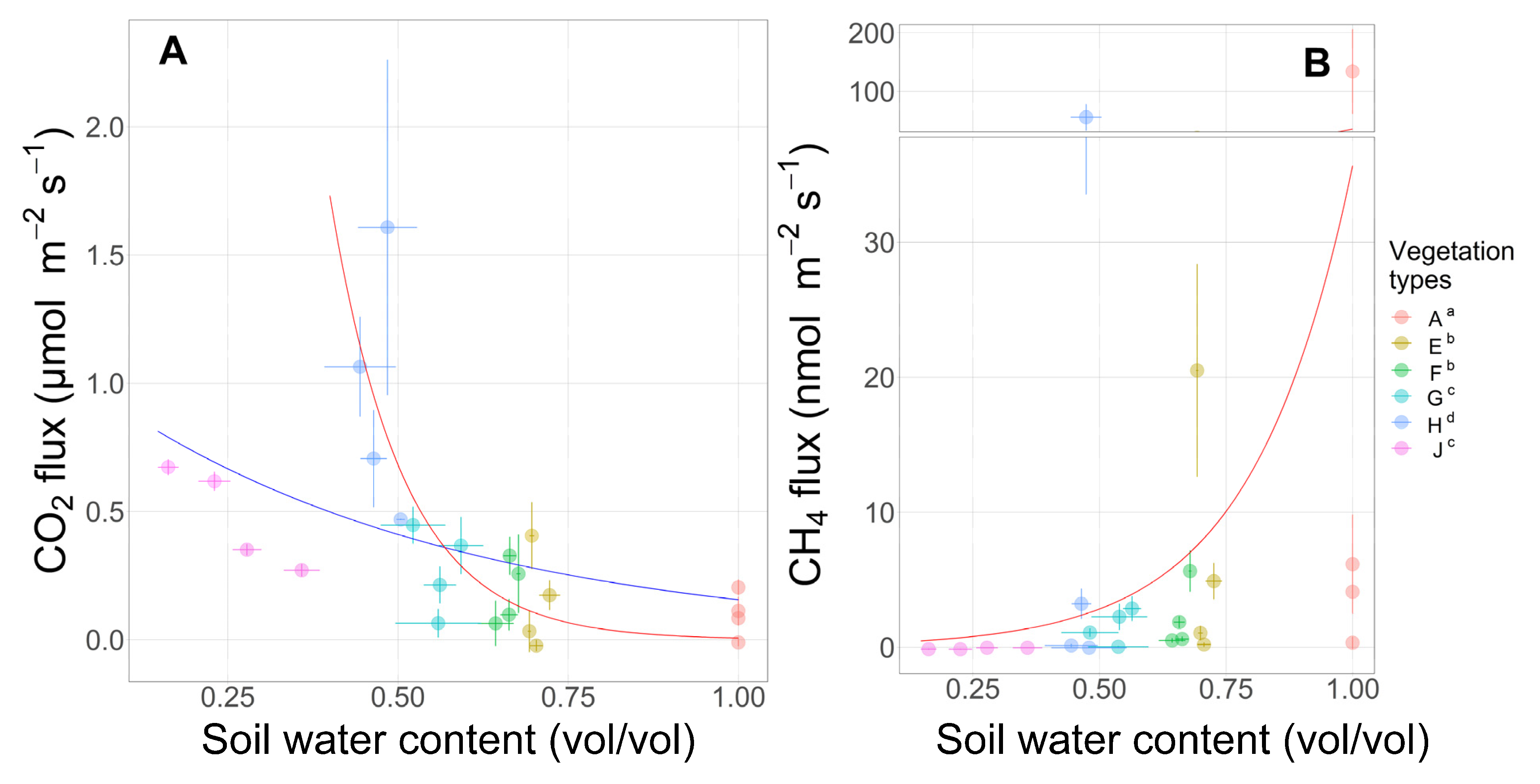
3.7. Surface CO2 and CH4 Fluxes
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baldrian, P. The known and the unknown in soil microbial ecology. FEMS Microbiol. Ecol. 2019, 95, fiz005. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.B.; Lajtha, K.; Crow, S.E.; Hugelius, G.; Kramer, M.G.; Piñeiro, G. The ecology of soil carbon: Pools, vulnerabilities, and biotic and abiotic controls. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 419–445. [Google Scholar] [CrossRef] [Green Version]
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2017, 41, 109–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldrian, P.; Kolařík, M.; Štursová, M.; Kopecký, J.; Valášková, V.; Větrovský, T.; Žifčáková, L.; Šnajdr, J.; Rídl, J.; Vlček, Č.; et al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012, 6, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Žifčáková, L.; Větrovský, T.; Howe, A.; Baldrian, P. Microbial activity in forest soil reflects the changes in ecosystem properties between summer and winter. Environ. Microbiol. 2016, 18, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef] [Green Version]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Drivers of microbial community structure in forest soils. Appl. Microbiol. Biotechnol. 2018, 102, 4331–4338. [Google Scholar] [CrossRef]
- Loisel, J.; Gallego-Sala, A.V.; Amesbury, M.J.; Magnan, G.; Anshari, G.; Beilman, D.W.; Benavides, J.C.; Blewett, J.; Camill, P.; Charman, D.J.; et al. Expert assessment of future vulnerability of the global peatland carbon sink. Nat. Clim. Chang. 2021, 11, 70–77. [Google Scholar] [CrossRef]
- Rydin, H.; Jeglum, J.K.; Bennett, K.D. The Biology of Peatlands, 2nd ed.; Oxford University Press: Oxford, UK, 2013. [Google Scholar] [CrossRef]
- Xu, J.; Morris, P.J.; Liu, J.; Holden, J. PEATMAP: Refining estimates of global peatland distribution based on a meta-analysis. Catena 2018, 160, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Loisel, J.; Brosseau, D.P.; Beilman, D.W.; Hunt, S.J. Global peatland dynamics since the Last Glacial Maximum. Geophys. Res. Lett. 2010, 37, L13402. [Google Scholar] [CrossRef]
- Kostka, J.E.; Weston, D.J.; Glass, J.B.; Lilleskov, E.A.; Shaw, A.J.; Turetsky, M.R. The Sphagnum microbiome: New insights from an ancient plant lineage. New Phytol. 2016, 211, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Dom, S.P.; Ikenaga, M.; Lau, S.Y.L.; Radu, S.; Midot, F.; Yap, M.L.; Chin, M.-Y.; Lo, M.L.; Jee, M.S.; Maie, N.; et al. Linking prokaryotic community composition to carbon biogeochemical cycling across a tropical peat dome in Sarawak, Malaysia. Sci. Rep. 2021, 11, 6416. [Google Scholar] [CrossRef] [PubMed]
- Bräuer, L.S.; Basiliko, N.; Siljanen, H.M.P.; Zinder, S.H. Methanogenic archaea in peatlands. FEMS Microbiol. Lett. 2020, 367, fnaa172. [Google Scholar] [CrossRef] [PubMed]
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; et al. The global methane budget 2000–2017. Earth Syst. Sci. Data 2020, 12, 1561–1623. [Google Scholar] [CrossRef]
- Zhang, Z.; Furman, A. Soil redox dynamics under dynamic hydrologic regimes-A review. Sci. Total Environ. 2021, 763, 143026. [Google Scholar] [CrossRef]
- Guerrero-Cruz, S.; Vaksmaa, A.; Horn, M.A.; Niemann, H.; Pijuan, M.; Ho, A. Methanotrophs: Discoveries, environmental relevance, and a perspective on current and future applications. Front. Microbiol. 2021, 12, 678057. [Google Scholar] [CrossRef]
- Chang, S.-C.; Schemenauer, R.S. Fog Deposition. In Springer Handbook of Atmospheric Measurements; Foken, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar] [CrossRef]
- Tropical Montane Cloud Forests: Science for Conservation and Management; Bruijnzeel, L.A.; Scatena, F.N.; Hamilton, L.S. (Eds.) Cambridge University Press: Cambridge, UK, 2010; p. 768. [Google Scholar]
- Jones, S.P.; Diem, T.; Huaraca Quispe, L.P.; Cahuana, A.J.; Reay, D.S.; Meir, P.; Teh, Y.A. Drivers of atmospheric methane uptake by montane forest soils in the southern Peruvian Andes. Biogeosciences 2016, 13, 4151–4165. [Google Scholar] [CrossRef] [Green Version]
- Schulz, H.M.; Li, C.F.; Thies, B.; Chang, S.C.; Bendix, J. Mapping the montane cloud forest of Taiwan using 12 year MODIS-derived ground fog frequency data. PLoS One 2017, 12, e0172663. [Google Scholar] [CrossRef]
- Beiderwieden, E.; Schmidt, A.; Hsia, Y.J.; Chang, S.C.; Wrzesinsky, T.; Klemm, O. Nutrient input through occult and wet deposition into a subtropical montane cloud forest. Water Air Soil Pollut. 2007, 186, 273–288. [Google Scholar] [CrossRef]
- Chang, S.C.; Yeh, C.F.; Wu, M.J.; Hsia, Y.J.; Wu, J.T. Quantifying fog water deposition by in situ exposure experiments in a mountainous coniferous forest in Taiwan. For. Ecol. Manag. 2006, 224, 11–18. [Google Scholar] [CrossRef]
- Rees, R.; Chang, S.C.; Wang, C.P.; Matzner, E. Release of nutrients and dissolved organic carbon during decomposition of Chamaecyparis obtusa var. formosana leaves in a mountain forest in Taiwan. J. Plant Nutr. Soil Sci. 2006, 169, 792–798. [Google Scholar]
- Chang, S.C.; Tseng, K.H.; Hsia, Y.J.; Wang, C.P.; Wu, J.T. Soil respiration in a subtropical montane cloud forest in Taiwan. Agric. For. Meteorol. 2008, 148, 788–798. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Huang, Y.-J.; Tang, S.-L.; Whitman, W.B.; Coleman, D.C.; Chiu, C.-Y. Bacterial community diversity in undisturbed perhumid montane forest soils in Taiwan. Microb. Ecol. 2010, 59, 369–378. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Jangid, K.; Whitman, W.B.; Coleman, D.C.; Chiu, C.Y. Soil bacterial communities in native and regenerated perhumid montane forests. Appl. Soil Ecol. 2011, 47, 111–118. [Google Scholar] [CrossRef]
- Chang, E.-H.; Tian, G.; Chiu, C.-Y. Soil microbial communities in natural and managed cloud montane forests. Forests 2017, 8, 33. [Google Scholar] [CrossRef]
- Lai, I.-L.; Chang, S.-C.; Lin, P.-H.; Chou, C.-H.; Wu, J.-T. Climatic characteristics of the subtropical mountainous cloud forest at the Yuanyang Lake Long-Term Ecological Research Site, Taiwan. Taiwania 2006, 51, 317–329. [Google Scholar]
- Tsai, J.W.; Kratz, T.K.; Hanson, P.C.; Wu, J.T.; Chang, W.Y.B.; Arzberger, P.W.; Lin, B.S.; Lin, F.P.; Chou, H.M.; Chiu, C.Y. Seasonal dynamics, typhoons and the regulation of lake metabolism in a subtropical humic lake. Freshw. Biol. 2008, 53, 1929–1941. [Google Scholar] [CrossRef]
- Chou, C.-H.; Chen, T.-Y.; Liao, C.-C.; Peng, C.-I. Long-term ecological research in the Yuanyang Lake forest ecosystem I. Vegetation composition and analysis. Bot. Bull. Acad. Sin. 2000, 41, 61–72. [Google Scholar]
- Li, H.-L.; Keng, H. Cupressaceae. In Flora of Taiwan, 2nd ed.; Huang, T.-C., Hsieh, C.-F., Keng, H., Shieh, W.-C., Tsai, J.-L., Eds.; Editorial Committee of the Flora of Taiwan: Taipei, Taiwan, 1994; Volume 1, pp. 586–595. [Google Scholar]
- Shiau, Y.-J.; Pai, C.-W.; Tsai, J.-W.; Liu, W.-C.; Yam, R.S.W.; Chang, S.-C.; Tang, S.-L.; Chiu, C.-Y. Characterization of phosphorus in a toposequence of subtropical perhumid forest soils facing a subalpine lake. Forests 2018, 9, 294. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.-C.; Chou, C.-H.; Wu, J.-T. Population structure and substrates of Taiwan yellow false cypress (Chamaecyparis obtusa var. formosana) in Yuanyang Lake Nature Reserve and nerby Szumakuszu, Taiwan. Taiwania 2003, 48, 6–12. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D. Reintroducing mothur: 10 years later. Appl. Environ. Microbiol. 2020, 86, e02343-19. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Oren, A.; Arahal, D.R.; Rosselló-Móra, R.; Sutcliffe, I.C.; Moore, E.R.B. Emendation of Rules 5b, 8, 15 and 22 of the International Code of Nomenclature of Prokaryotes to include the rank of phylum. Int. J. Syst. Evol. Microbiol. 2021, 71. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.J.; Wrighton, K.C. Metagenomic approaches unearth methanotroph phylogenetic and metabolic diversity. Curr. Issues Mol. Biol. 2019, 33, 57–84. [Google Scholar] [CrossRef] [Green Version]
- Leys, C.; Ley, C.; Klein, O.; Bernard, P.; Licata, L. Detecting outliers: Do not use standard deviation around the mean, use absolute deviation around the median. J. Exp. Soc. Psychol. 2013, 49, 764–766. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 19 May 2022).
- Hannula, S.E.; Heinen, R.; Huberty, M.; Steinauer, K.; De Long, J.R.; Jongen, R.; Bezemer, T.M. Persistence of plant-mediated microbial soil legacy effects in soil and inside roots. Nat. Commun. 2021, 12, 5686. [Google Scholar] [CrossRef] [PubMed]
- Wicaksono, W.A.; Cernava, T.; Berg, C.; Berg, G. Bog ecosystems as a playground for plant-microbe coevolution: Bryophytes and vascular plants harbour functionally adapted bacteria. Microbiome 2021, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Kitson, E.; Bell, N.G.A. The response of microbial communities to peatland drainage and rewetting. A review. Front. Microbiol. 2020, 11, 582812. [Google Scholar] [CrossRef]
- Imberger, K.T.; Chiu, C.-Y. Topographical and seasonal effects on soil fungal and bacterial activity in subtropical, perhumid, primary and regenerated montane forests. Soil Biol. Biochem. 2002, 34, 711–720. [Google Scholar] [CrossRef]
- Bledsoe, R.B.; Goodwillie, C.; Peralta, A.L. Long-term nutrient enrichment of an oligotroph-dominated wetland increases bacterial diversity in bulk soils and plant rhizospheres. mSphere 2020, 5, e.00035-20. [Google Scholar] [CrossRef]
- Lamit, L.J.; Romanowicz, K.J.; Potvin, L.R.; Lennon, J.T.; Tringe, S.G.; Chimner, R.A.; Kolka, R.K.; Kane, E.S.; Lilleskov, E.A. Peatland microbial community responses to plant functional group and drought are depth-dependent. Mol. Ecol. 2021, 30, 5119–5136. [Google Scholar] [CrossRef]
- Rupp, D.L.; Lamit, L.J.; Techtmann, S.M.; Kane, E.S.; Lilleskov, E.A.; Turetsky, M.R. The rhizosphere responds: Rich fen peat and root microbial ecology after long-term water table manipulation. Appl. Environ. Microbiol. 2021, 87, e0024121. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Wegner, C.E.; Kim, Y.; Liesack, W.; Dedysh, S.N. Identification of microbial populations driving biopolymer degradation in acidic peatlands by metatranscriptomic analysis. Mol. Ecol. 2016, 25, 4818–4835. [Google Scholar] [CrossRef]
- Kaupper, T.; Mendes, L.W.; Poehlein, A.; Frohloff, D.; Rohrbach, S.; Horn, M.A.; Ho, A. The methane-driven interaction network in terrestrial methane hotspots. Environ. Microbiome 2022, 17, 15. [Google Scholar] [CrossRef]
- Chang, S.-C.; Wang, C.-P.; Feng, C.-M.; Rees, R.; Hell, U.; Matzner, E. Soil fluxes of mineral elements and dissolved organic matter following manipulation of leaf litter input in a Taiwan Chamaecyparis forest. For. Ecol. Manag. 2007, 242, 133–141. [Google Scholar] [CrossRef]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The ecology of acidobacteria: Moving beyond genes and genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalam, S.; Basu, A.; Ahmad, I.; Sayyed, R.Z.; El-Enshasy, H.A.; Dailin, D.J.; Suriani, N.L. Recent understanding of soil acidobacteria and their ecological significance: A critical review. Front. Microbiol. 2020, 11, 580024. [Google Scholar] [CrossRef] [PubMed]
- Foesel, B.U.; Mayer, S.; Luckner, M.; Wanner, G.; Rohde, M.; Overmann, J. Occallatibacter riparius gen. nov., sp. nov. and Occallatibacter savannae sp. nov., acidobacteria isolated from Namibian soils, and emended description of the family Acidobacteriaceae. Int. J. Syst. Evol. Microbiol. 2016, 66, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Dedysh, S.N.; Ivanova, A.A. Planctomycetes in boreal and subarctic wetlands: Diversity patterns and potential ecological functions. FEMS Microbiol. Ecol. 2019, 95, fiy227. [Google Scholar] [CrossRef] [Green Version]
- Sockett, R.E. Predatory lifestyle of Bdellovibrio bacteriovorus. Annu. Rev. Microbiol. 2009, 63, 523–539. [Google Scholar] [CrossRef]
- Soo, R.M.; Woodcroft, B.J.; Parks, D.H.; Tyson, G.W.; Hugenholtz, P. Back from the dead; the curious tale of the predatory cyanobacterium Vampirovibrio chlorellavorus. PeerJ 2015, 3, e968. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-N.; Lee, T.-Q.; Meyers, P.A.; Fan, C.-W.; Chen, R.-F.; Wei, K.-Y.; Chen, Y.-G.; Wu, J.-T. The effect of typhoon induced rainfall on settling fluxes of particles and organic carbon in Yuanyang Lake, subtropical Taiwan. J. Asian Earth Sci. 2011, 40, 1171–1179. [Google Scholar] [CrossRef]
- Huang, Y.; Ciais, P.; Luo, Y.; Zhu, D.; Wang, Y.; Qiu, C.; Goll, D.S.; Guenet, B.; Makowski, D.; De Graaf, I.; et al. Tradeoff of CO2 and CH4 emissions from global peatlands under water-table drawdown. Nat. Clim. Chang. 2021, 11, 618–622. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vegetation Type | A | B | C | D | E | F | G | H | I * | J |
|---|---|---|---|---|---|---|---|---|---|---|
| Ecosystem | Peatland | Forest | ||||||||
| Physiognomic group | Marsh | Bog | Thicket swamp forest | Conifer swamp forest | Upland in watershed | Outside watershed | ||||
| Water level (cm) | 46.0 | 36.8 | 2.5 | 2.8 | 7.1 | 2.0 | 0 | 0 | 0 | 0 |
| Plant species number | 1 | 1 | 5 | 4 | 5 | 4 | 14 | 6 | - | 15 |
| Wetland plant species | 1 | 1 | 3 | 3 | 4 | 3 | 3 | 0 | 0 | 0 |
| Percent wetland species | 100 | 100 | 60 | 75 | 80 | 75 | 21.4 | 0 | 0 | 0 |
| Herbaceous species number | 1 | 1 | 5 | 4 | 5 | 4 | 7 | 4 | - | 5 |
| Percent herbaceous species number | 100 | 100 | 100 | 100 | 100 | 100 | 50 | 67 | - | 33 |
| Coverage of wetland species (%) | 84 | 82 | 78 | 28 | 90 | 74 | 25 | 0 | 0 | 0 |
| Coverage of herbaceous species (%) | 84 | 82 | 100 | 100 | 100 | 100 | 100 | 3 | - | 23 |
| Mosses and lichen (% ground area) | 0 | 0 | 19.7 | 19.3 | 65.7 | 22.7 | 41.0 | 30.0 | - | 83.0 |
| Woody species number | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 2 | 42 | 10 |
| Woody species density (individuals ha−1) | 9333 | 1200 | 6688 | 2367 | ||||||
| Woody species basal area (m2 ha−1) | 0 | 0 | 0 | 0 | 0 | 0 | 18.4 | 86.3 | 58.6 | 54.5 |
| Yellow cypress density (individuals ha−1) | 0 | 300 | 848 | 1367 | ||||||
| Individual yellow cypress basal area (cm2) | 0 | 1667 | 480 | 360 | ||||||
| Microbial sampling plot | F | G | Ha,b,c | I | Ja, b | |||||
| CO2 and CH4 flux sampling plot | A | E | F | G | H | J | ||||
| Life Form | Species | A | B | C | D | E | F | G | H | I * | J |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Woody species | |||||||||||
| Chamaecyparis obtusa var. formosana | 50.0 | 40.7 | 49.0 | ||||||||
| Rhododendron formosanum | 36.3 | 9.8 | |||||||||
| Illicium anisatum | 3.5 | 0.13 | |||||||||
| Adinandra formosana var. formosana | 1.1 | ||||||||||
| Schefflera taiwaniana | 0.3 | ||||||||||
| Barthea barthei | 0.2 | ||||||||||
| Viburnum sympodiale | 0.1 | ||||||||||
| Dendropanax dentiger | 0.7 | ||||||||||
| Skimmia arisanensis | 0.04 | ||||||||||
| Ternstroemia gymnanthera | 0.8 | ||||||||||
| Neolitsea acuminatissima | 0.1 | ||||||||||
| Hydrangea paniculata | 14.02 | ||||||||||
| Rhododendron chilanshanense | 2.51 | ||||||||||
| Prunus campanulata | 1.97 | ||||||||||
| Acer serrulatum | 1.38 | ||||||||||
| Lindera communis | 1.28 | ||||||||||
| Dendropanax dentiger | 0.1 | 1.0 | |||||||||
| Vaccinium wrightii var. wrightii | 0.47 | ||||||||||
| Ilex hayatana | 0.3 | ||||||||||
| Rhamnus nakaharae | 0.2 | ||||||||||
| Eurya crenatifolia | 0.09 | 0.06 | |||||||||
| Machilus thunbergia | 0.14 | ||||||||||
| Cleyera japonica var. japonica | 0.1 | ||||||||||
| Lyonia ovalifolia var. ovalifolia | 0.04 | ||||||||||
| Viburnum luzonicum | 0.01 | ||||||||||
| Herbaceous species | |||||||||||
| Aquatic | Schoenoplectus mucronatus ssp. robustus | 83.7 | 88.3 | 4.3 | 21.8 | ||||||
| Trichophorum subcapitatum | 28.0 | 3.2 | 78.0 | 21.3 | |||||||
| Rhynchospora alba | 81.8 | 0.7 | |||||||||
| Sparganium fallax | 82.3 | ||||||||||
| Juncus effusus var. decipiens | 6.0 | 2.3 | 7.0 | 4.3 | |||||||
| Persicaria sagittate | 15.3 | ||||||||||
| Persicaria thunbergii | 4.3 | 0.3 | |||||||||
| Terrestrial | Miscanthus transmorrisonensis | 21.3 | 100 | 11.5 | 29.3 | 76.7 | |||||
| Plagiogyria glauca | 2 | 12 | |||||||||
| Viola adenothrix var. adenothrix | 9.0 | ||||||||||
| Elatostema trilobulatum | 6 | ||||||||||
| Ophiopogon intermedius | 3 | ||||||||||
| Hymenophyllum paniculiflorum | 0.33 | 2 | |||||||||
| Sarcopyramis napalensis var. delicata | 0.33 | 1 | |||||||||
| Tripterospermum lanceolatum | 0.33 | <0.01 | |||||||||
| Rubus corchorifolius | 0.33 | <0.01 | |||||||||
| Nertera nigricarpa | 0.33 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Lai, I.-L.; Chang, S.-C. Changes in Soil Microbial Community and Carbon Flux Regime across a Subtropical Montane Peatland-to-Forest Successional Series in Taiwan. Forests 2022, 13, 958. https://doi.org/10.3390/f13060958
Chen C-Y, Lai I-L, Chang S-C. Changes in Soil Microbial Community and Carbon Flux Regime across a Subtropical Montane Peatland-to-Forest Successional Series in Taiwan. Forests. 2022; 13(6):958. https://doi.org/10.3390/f13060958
Chicago/Turabian StyleChen, Chun-Yao, I-Ling Lai, and Shih-Chieh Chang. 2022. "Changes in Soil Microbial Community and Carbon Flux Regime across a Subtropical Montane Peatland-to-Forest Successional Series in Taiwan" Forests 13, no. 6: 958. https://doi.org/10.3390/f13060958
APA StyleChen, C. -Y., Lai, I. -L., & Chang, S. -C. (2022). Changes in Soil Microbial Community and Carbon Flux Regime across a Subtropical Montane Peatland-to-Forest Successional Series in Taiwan. Forests, 13(6), 958. https://doi.org/10.3390/f13060958