
Comprehensive Analysis of the DNA Methyltransferase Genes and Their Association with Salt Response in Pyrus betulaefolia

Abstract
:1. Introduction
2. Material and Methods
2.1. Identification of the MTase Genes
2.2. Sequence Analysis and Phylogenetic Tree Construction
2.3. Divergence Analysis and Ks Calculation
2.4. Stress Treatments and Plant Materials Collection
2.5. Quantitative Real-Time PCR
2.6. Methylation Detection of Candidate Salt-Responsive Genes
3. Results
3.1. Identification and Comprehensive Analysis of PbeMTase Genes
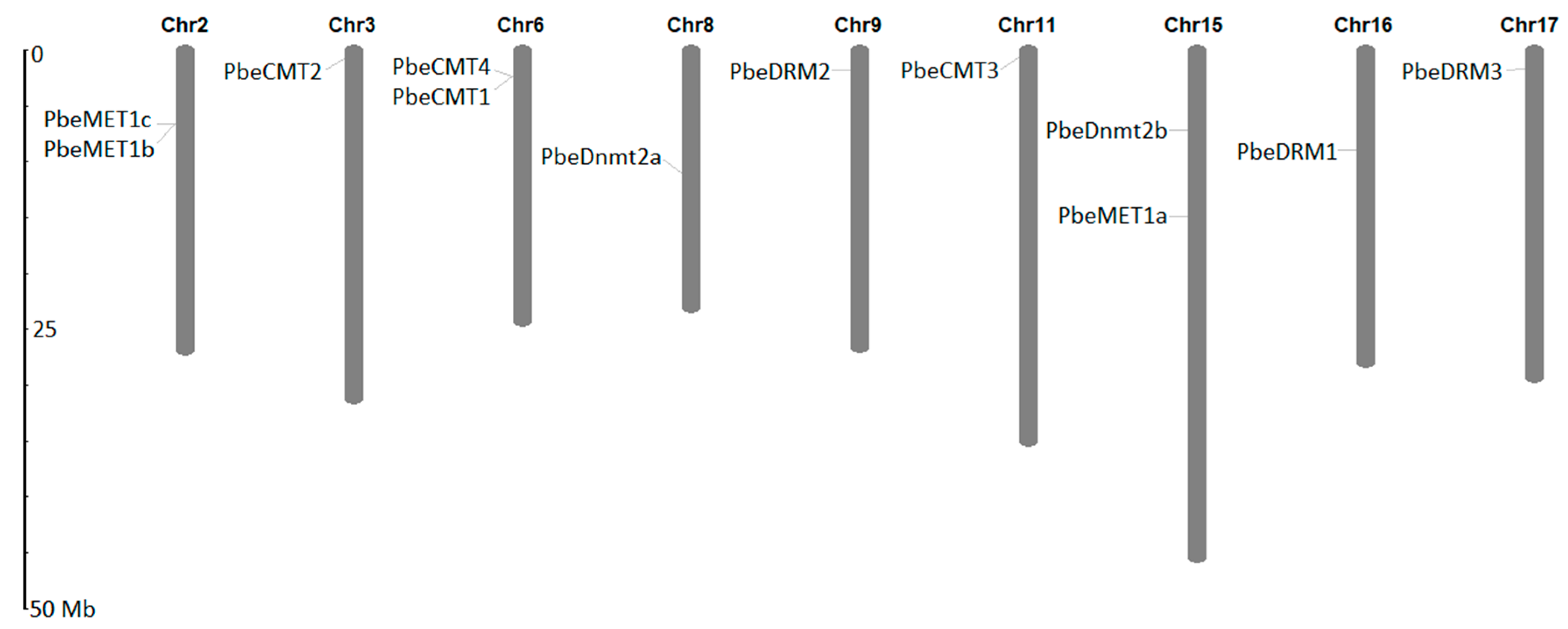
3.2. Gene Structure, Conserved Motif and Genomic Location Analysis of PbeMTases
3.3. Classification, Organization and Phylogeny of the PbeMTases
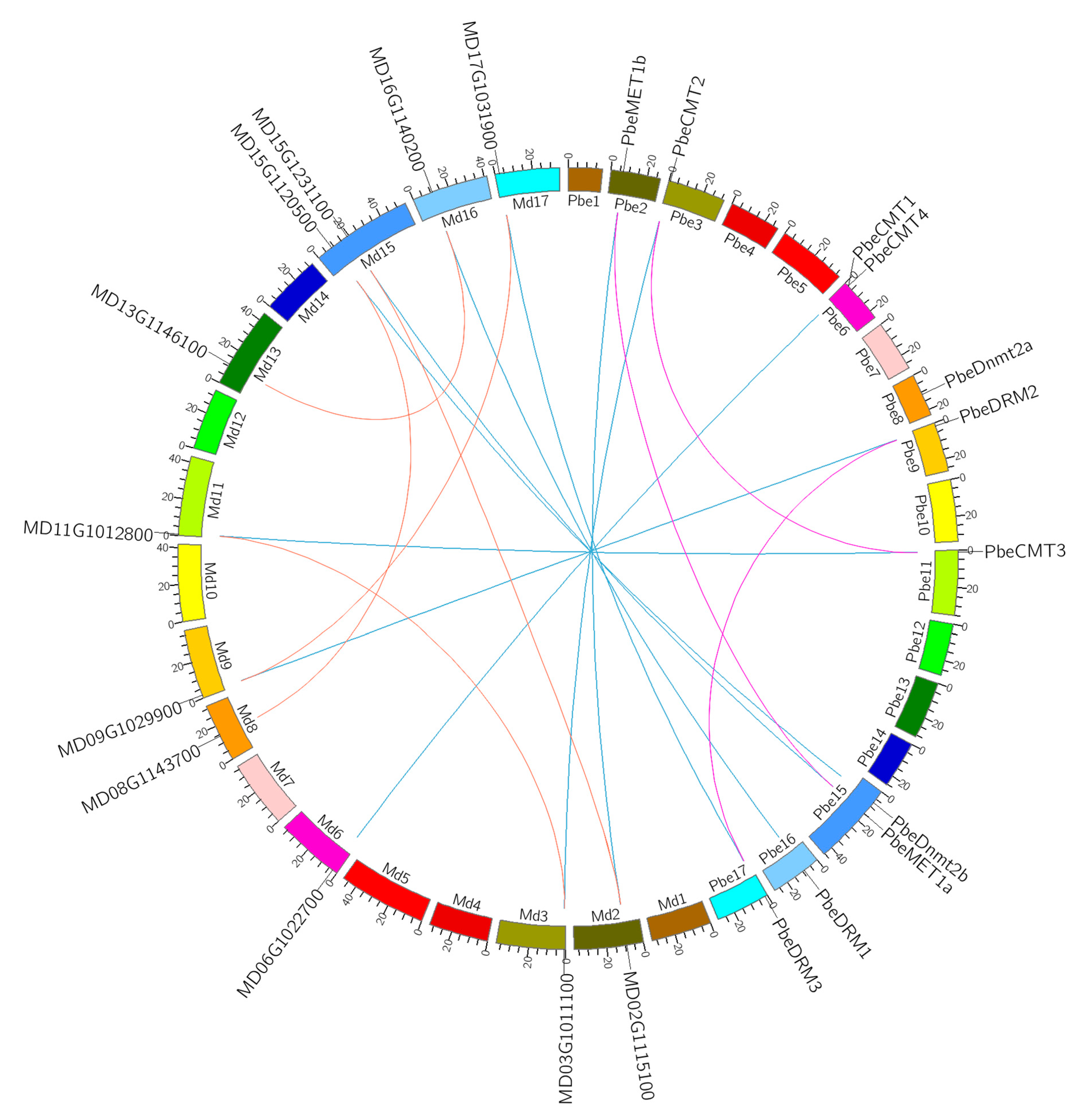
3.4. Evolutionary Clues of MTase Genes in P. betulaefolia and Apple
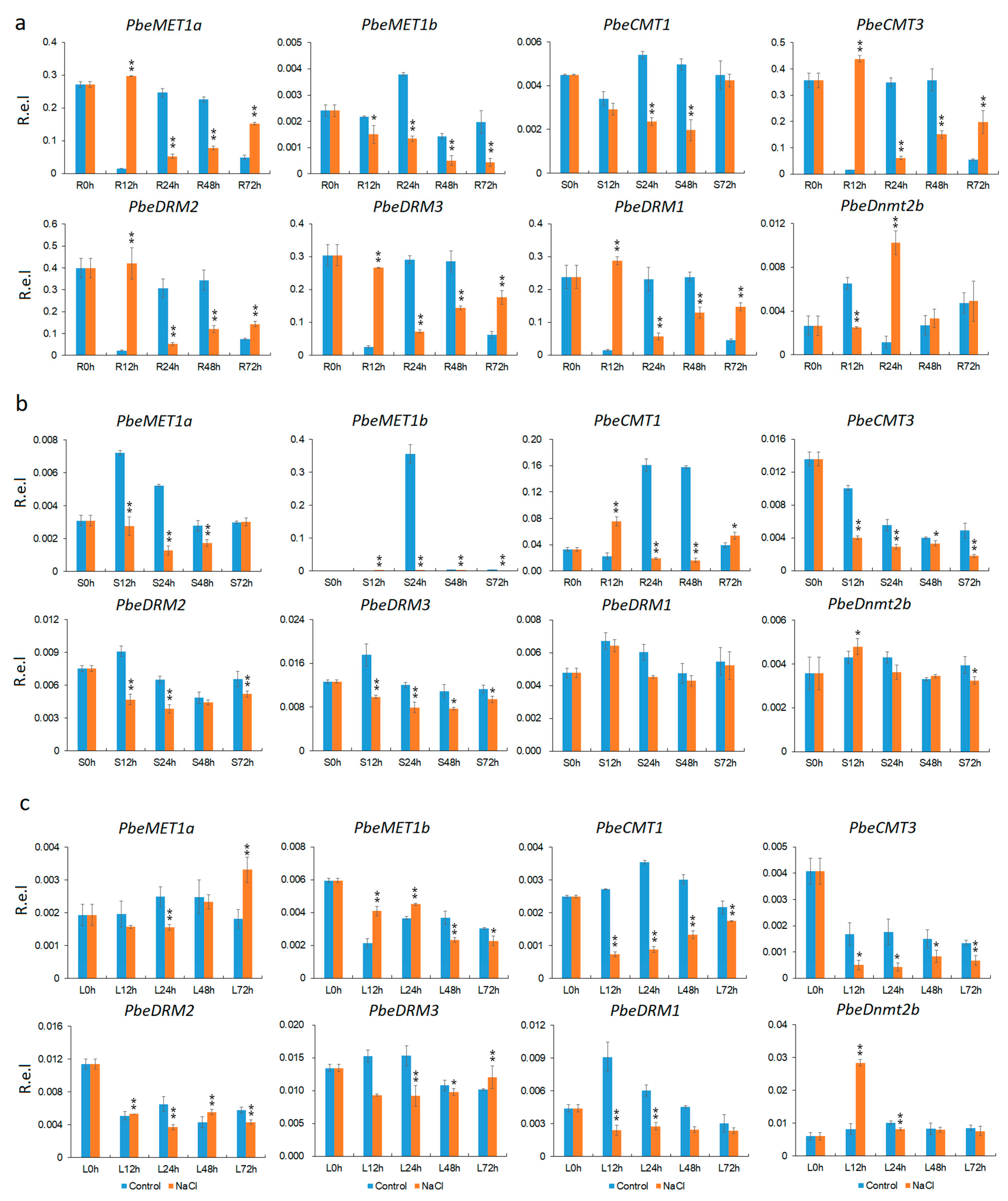
3.5. Tissue and Salt-Stress Expression Analysis
3.6. Methylation Level Changed of Salt-Responsive Genes under Salt Stress
4. Discussion
4.1. Conserved Gene Structures May Indicate Specific Functions
4.2. Roots Are a Key Organ for Plant Response to the Environment
4.3. Expression Divergence among Different Group Members of MTases
4.4. Conserved Evolution of PbeMTase Genes
4.5. Excellent Candidates for P. betulaefolia Salt Tolerance Breeding and Improvement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Holliday, R. Epigenetics: A historical overview. Epigenetics 2006, 1, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Martín-Subero, J.I. How epigenomics brings phenotype into being. Pediatr. Endocrinol. Rev. 2011, 9, 506–510. [Google Scholar] [PubMed]
- Yanjun, A.; Shenglin, J.; Zhengnan, C.; Botao, S.; Conghua, X.; Jun, L.; Jun, Z. DNA methylation affects photoperiodic tuberization in potato (Solanum tuberosum L.) by mediating the expression of genes related to the photoperiod and GA pathways. Hortic. Res. 2021, 8, 181. [Google Scholar]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [PubMed]
- María-Estefanía, L.; David, R.; Claude, B.; Béatrice, D.; Etienne, B. DNA methylation dynamics during stress response in woodland strawberry (Fragaria vesca). Hortic. Res. 2022, 9, uhac174. [Google Scholar]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Noy-Malka, C.; Yaari, R.; Itzhaki, R.; Mosquna, A.; Auerbach Gershovitz, N.; Katz, A.; Ohad, N. A single CMT methyltransferase homolog is involved in CHG DNA methylation and development of Physcomitrella patens. Plant. Mol. Biol. 2014, 84, 719–773. [Google Scholar] [CrossRef]
- Garg, R.; Kumari, R.; Tiwari, S.; Goyal, S. Genomic survey, gene expression analysis and structural modeling suggest diverse roles of DNA methyltransferases in legumes. PLoS ONE 2014, 9, e88947. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Kossida, S. Plant cytosine-5 DNA methyltransferases structure, function, and molecular evolution. Genomics 2007, 90, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7 Molecular Evolutionary Genetics Analysis version 70 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Na, E.S.; Autry, A.E.; Monteggia, L.M. Impact of DNMT1 and DNMT3a forebrain knockout on depressive- and anxiety like behavior in mice. Neurobiol. Learn. Mem. 2016, 135, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 9316, 8449–8454. [Google Scholar] [CrossRef]
- Steward, N.; Kusano, T.; Sano, H. Expression of ZmMET1, a gene encoding a DNA methyltransferase from maize, is associated not only with DNA replication in actively proliferating cells, but also with altered DNA methylation status in cold-stressed quiescent cells. Nucleic Acids. Res. 2000, 28, 3250–3259. [Google Scholar] [CrossRef] [PubMed]
- Teerawanichpan, P.; Chandrasekharan, M.; Jiang, Y.; Narangajavana, J.; Hall, T. Characterization of two rice DNA methyltransferase genes and RNA imediated reactivation of a silenced transgene in rice callus. Planta 2004, 218, 337–349. [Google Scholar]
- Fujimoto, R.; Sasaki, T.; Nishio, T. Characterization of DNA methyltransferase genes in Brassica rapa. Genes Genet. Syst. 2006, 81, 235–242. [Google Scholar] [CrossRef]
- Thomas, M.; Pingault, L.; Poulet, A.; Duarte, J.; Throude, M.; Faure, S.; Pichon, J.P.; Paux, E.; Probst, A.V.; Tatout, C. Evolutionary history of Methyltransferase 1 genes in hexaploid wheat. BMC Genom. 2014, 15, 922. [Google Scholar] [CrossRef]
- Kim, M.; Ohr, H.; Lee, J.W.; Hyun, Y.; Fischer, R.L.; Choi, Y. Temporal and spatial downregulation of Arabidopsis MET1 activity results in global DNA hypomethylation and developmental defects. Mol. Cells 2008, 26, 611–615. [Google Scholar]
- Cao, X.F.; Jacobsen, S.E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 2002, 994, 16491–16498. [Google Scholar] [CrossRef]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–995. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Luo, Z.; Zhao, X.; Li, S.; Wu, F.; Chen, J.; Xiang, M. Exogenously applied methyl jasmonate increased the resistance of postharvest pear fruit to blue mold. Fruit Res. 2022, 2, 11. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Z.; Shi, Z.; Zhang, S.; Ming, R.; Zhu, S.; Khan, M.A.; Tao, S.; Korban, S.S.; Wang, H.; et al. The genome of the pear (Pyrus bretschneideri Rehd). Genome Res. 2013, 3, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yuan, H.; Yang, Q.; Li, M.; Wang, Y.; Li, Y.; Ma, X.; Tan, F.; Wu, R. The genetic architecture of growth traits in Salix matsudana under salt stress. Hortic. Res. 2017, 4, 17024. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Cao, H.; Wang, Q.; Niu, Y.; Sadeghnezhad, E.; Han, S.; Liu, M.; Wang, L.; Liu, Z. Transcriptome analysis revealed MAPK and hormone pathway involving in exogenous melatonin-regulated salt tolerance in sour jujube. Fruit Res. 2023, 3, 19. [Google Scholar] [CrossRef]
- Dong, X.; Wang, Z.; Tian, L.; Zhang, Y.; Qi, D.; Huo, H.; Xu, J.; Li, Z.; Liao, R.; Shi, M.; et al. De novo assembly of a wild pear (Pyrus betuleafolia) genome. Plant Biotechnol. J. 2019, 18, 581–595. [Google Scholar] [CrossRef]
- Matsumoto, K.; Tamura, F.; Chun, J.P.; Ikeda, T.; Imanishi, K.; Tanabe, K. Enhancement in salt tolerance of Japanese pear by using Pyrus betulaefolia rootstock. Hortic. Res. 2007, 6, 47–52. [Google Scholar] [CrossRef]
- Zhang, X.; Lii, Y.; Wu, Z.; Polishko, A.; Zhang, H.; Chinnusamy, V.; Lonardi, S.; Zhu, J.K.; Liu, R.; Jin, H. Mechanisms of small RNA generation from cis-NATs in response to environmental and developmental cues. Mol. Plant 2013, 6, 704–715. [Google Scholar] [CrossRef]
- Wang, M.; Qin, L.; Xie, C.; Li, W.; Yuan, J.; Kong, L.; Yu, W.; Xia, G.; Liu, S. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol. 2014, 55, 1354–1365. [Google Scholar] [CrossRef]
- Ferreira, L.J.; Azevedo, V.; Maroco, J.; Oliveira, M.M.; Santos, A.P. Salt tolerant and sensitive rice varieties display differential methylome flexibility under salt stress. PLoS ONE 2015, 10, e0124060. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server interactive sequence similarity searching. Nucleic. Acids. Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Huang, J.; Wang, H.; Xie, X.; Zhang, D.; Liu, Y.; Guo, G. Roles of DNA methyltransferases in Arabidopsis development. Afr. J. Biotechnol. 2010, 9, 8506–8514. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 20. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Cheng, X.; Klimasauskas, S.; Mi, S.; Posfai, J.; Roberts, R.J.; Wilson, G.G. The DNA (cytosine-5) methyltransferases. Nucleic. Acids. Res. 1994, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5 a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos an information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Ma, N.; Liu, C.; Li, H.; Wang, J.; Zhang, B.; Lin, J.; Chang, Y. Genome-wide identification of lectin receptor kinases in pear, Functional characterization of the L-type LecRLK gene PbLRK138. Gene 2018, 661, 11–21. [Google Scholar] [CrossRef]
- Hao, X.; Horvath, D.P.; Chao, W.S.; Yang, Y.; Wang, X.; Xiao, B. Identification and Evaluation of Reliable Reference Genes for Quantitative Real-Time PCR Analysis in Tea Plant (Camellia sinensis (L.) O. Kuntze). Int. J. Mol. Sci. 2014, 15, 22155–22172. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.R.; Deleris, A.; Wong, W.; Zhong, X.; Chin, H.G.; Horwitz, G.A.; Kelly, K.A.; Pradhan, S.; Jacobsen, S.E. The de novo cytosine methyltransferase DRM2 requires intact UBA domains and a catalytically mutated paralog DRM3 during RNA directed DNA methylation in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1001182. [Google Scholar] [CrossRef]
- Wang, Y.N.; Li, K.X.; Li, X. Auxin redistribution modulates plastic development of root system architecture under salt stress in Arabidopsis thaliana. J. Plant. Physiol. 2009, 166, 1637–1645. [Google Scholar] [CrossRef]
- Meng, D.; Fricke, W. Changes in root hydraulic conductivity facilitate the overall hydraulic response of rice (Oryza sativa L.) cultivars to salt and osmotic stress. Plant. Physiol. Biochem. 2017, 113, 64–77. [Google Scholar] [CrossRef]
- Mayak, S.; Tirosh, T.; Glick, B.R. Plant growth-promoting bacteria confer resistance in tomato plants to salt stress. Plant. Physiol. Biochem. 2004, 42, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, K.; Ehsanpour, A.A.; Shah, A.H.; Komatsu, S. Proteome analysis of soybean hypocotyl and root under salt stress. Amino Acids. 2009, 36, 91–98. [Google Scholar] [CrossRef]
- Rossi, L.; Zhang, W.; Ma, X. Cerium oxide nanoparticles alter the salt stress tolerance of Brassica napus L. by modifying the formation of root apoplastic barriers. Environ. Pollut. 2017, 229, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, I.; White, J.F. Application of bacteria from non-cultivated plants to promote growth, alter root architecture and alleviate salt stress of cotton. J. Appl. Microbiol. 2017, 122, 1110–1120. [Google Scholar] [CrossRef]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P.; et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell 2013, 25, 2783–2797. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xia, H.; Zhang, Y.; Zhao, S.; Zhao, C.; Hou, L.; Li, C.; Li, A.; Ma, C.; Wang, X. Genome-wide high resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in Maize (Zea may). BMC Genom. 2015, 16, 21. [Google Scholar] [CrossRef]
- Heard, E.; Disteche, C.M. Dosage compensation in mammals finetuning the expression of the X chromosome. Genes 2006, 20, 1848–1867. [Google Scholar] [CrossRef]
- Birchler, J.A.; Bhadra, U.; Bhadra, M.P.; Auger, D.L. Dosage-dependent gene regulation in multicellular eukaryotes implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev. Biol. 2001, 234, 275–288. [Google Scholar] [CrossRef]
- Deleris, A.; Halter, T.; Navarro, L. DNA Methylation and Demethylation in Plant Immunity. Annu. Rev. Phytopathol. 2016, 54, 579–603. [Google Scholar] [CrossRef]
- Li, X.L.; Lin, Z.X.; Nie, Y.C.; Guo, X.P.; Zhang, X.L. MSAP analysis of epigenetic changes in cotton (Gossypium hirsutum L.) under salt stress. Acta Agronom. Sin. 2009, 35, 588–596. [Google Scholar] [CrossRef]
- Yaish, M.W.; Abbas, A.L.; Ibtisam, A.H.; Vishwas, P.H. Genome-wide dna methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genom. 2018, 19, 78. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, F.; Qin, Q.; Zhao, X.; Li, Z.; Fu, B. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem. Biophys. Res. Commun. 2015, 465, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Al-Lawati, A.; Al-Bahry, S.; Victor, R.; Al-Lawati, A.H.; Yaish, M.W. Salt stress alters dna methylation levels in alfalfa (Medicago spp.). Genet. Mol. Res. 2016, 15, 15018299. [Google Scholar] [CrossRef]
- Bharti, P.; Mahajan, M.; Vishwakatma, A.K.; Bhardwaj, J.; Yadav, S.K. Atros1 overexpression provides evidence for epigenetic regulation of genes encoding enzymes of flavonoid biosynthesis and antioxidant pathways during salt stress in transgenic tobacco. J. Exp. Bot. 2015, 66, 5959–5969. [Google Scholar] [CrossRef]
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Zheng, C. Salt-induced transcription factor myb74 is regulated by the rna-directed dna methylation pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; et al.; International Peach Genome Initiative The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, T. Expansion and stress responses of the AP2/EREBP superfamily in cotton. BMC Genom. 2017, 18, 118. [Google Scholar] [CrossRef]
- Riddle, N.C.; Birchler, J.A. Effects of reunited diverged regulatory hierarchies in allopolyploids and species hybrids. Trends Genet. 2003, 19, 597–600. [Google Scholar] [CrossRef]
- Garg, R.; Chevala, V.N.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Science 2015, 5, 144922. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Li, M.; Lou, Q.; Xia, H.; Wang, P.; Li, T.; Liu, H.; Luo, L. Transgenerational variations in DNA methylation induced by drought stress in two rice varieties with distinguished difference to drought resistance. PLoS ONE 2013, 8, e80253. [Google Scholar] [CrossRef]
- Sun, L.; Miao, X.; Cui, J.; Deng, J.; Wang, X.; Wang, Y.; Zhang, Y.; Gao, S.; Yang, K. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across different salt stress in Maize (Zea mays L). Euphytica 2018, 214, 25. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Flowers, T.J.; Colmer, T.D. Salinity tolerance in halophytes. New Phytol. 2008, 179, 945–963. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Position | No. of Intron | CDS (bp) | Size (aa) | MW (kDa) | PI |
|---|---|---|---|---|---|---|---|
| PbeMET1a | GWHPAAYT021621 | Chr15:14910721-14914680 (+) | 1 | 3876 | 1291 | 145.03 | 5.48 |
| PbeMET1b | GWHPAAYT032029 | Chr2:6616009-6621452 (+) | 2 | 4686 | 1561 | 174.32 | 5.66 |
| PbeMET1c | GWHPAAYT032026 | Chr2:6586996-6589839 (+) | 0 | 2844 | 947 | 105.72 | 5.68 |
| PbeCMT1 | GWHPAAYT044647 | Chr6:2315152-2324586 (−) | 20 | 2493 | 830 | 94.88 | 6.33 |
| PbeCMT2 | GWHPAAYT034309 | Chr3:750623-756027 (−) | 20 | 2571 | 856 | 96.45 | 4.94 |
| PbeCMT3 | GWHPAAYT006172 | Chr11:703571-710209 (−) | 20 | 3159 | 1052 | 118.24 | 5.52 |
| PbeCMT4 | GWHPAAYT044646 | Chr6:2305737-2313279 (−) | 16 | 1881 | 626 | 71.36 | 7.24 |
| PbeDRM1 | GWHPAAYT025788 | Chr16:8967099-8972756 (+) | 11 | 2124 | 707 | 80.14 | 5.32 |
| PbeDRM2 | GWHPAAYT053650 | Chr9:1746585-1754810 (−) | 22 | 3264 | 1087 | 122.90 | 5.58 |
| PbeDRM3 | GWHPAAYT028091 | Chr17:1656828-1660671 (−) | 8 | 1803 | 600 | 67.64 | 4.74 |
| PbeDnmt2a | GWHPAAYT052082 | Chr8:10976646-10978580 (−) | 9 | 945 | 314 | 35.99 | 5.82 |
| PbeDnmt2b | GWHPAAYT020534 | Chr15:7135328-7152877 (−) | 17 | 2946 | 981 | 110.66 | 6.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Liu, C.; Xu, X.; Kan, J.; Li, H.; Lin, J.; Cheng, Z.; Chang, Y. Comprehensive Analysis of the DNA Methyltransferase Genes and Their Association with Salt Response in Pyrus betulaefolia. Forests 2023, 14, 1751. https://doi.org/10.3390/f14091751
Zhang Y, Liu C, Xu X, Kan J, Li H, Lin J, Cheng Z, Chang Y. Comprehensive Analysis of the DNA Methyltransferase Genes and Their Association with Salt Response in Pyrus betulaefolia. Forests. 2023; 14(9):1751. https://doi.org/10.3390/f14091751
Chicago/Turabian StyleZhang, Yufeng, Chunxiao Liu, Xiaoyang Xu, Jialiang Kan, Hui Li, Jing Lin, Zongming Cheng, and Youhong Chang. 2023. "Comprehensive Analysis of the DNA Methyltransferase Genes and Their Association with Salt Response in Pyrus betulaefolia" Forests 14, no. 9: 1751. https://doi.org/10.3390/f14091751
APA StyleZhang, Y., Liu, C., Xu, X., Kan, J., Li, H., Lin, J., Cheng, Z., & Chang, Y. (2023). Comprehensive Analysis of the DNA Methyltransferase Genes and Their Association with Salt Response in Pyrus betulaefolia. Forests, 14(9), 1751. https://doi.org/10.3390/f14091751