Immune Exhaustion: Past Lessons and New Insights from Lymphocytic Choriomeningitis Virus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Lymphocytic Choriomeningitis Virus (LCMV) and the Definition of T Cell Exhaustion
2. The Properties and Molecular Regulators of Exhausted T Cells
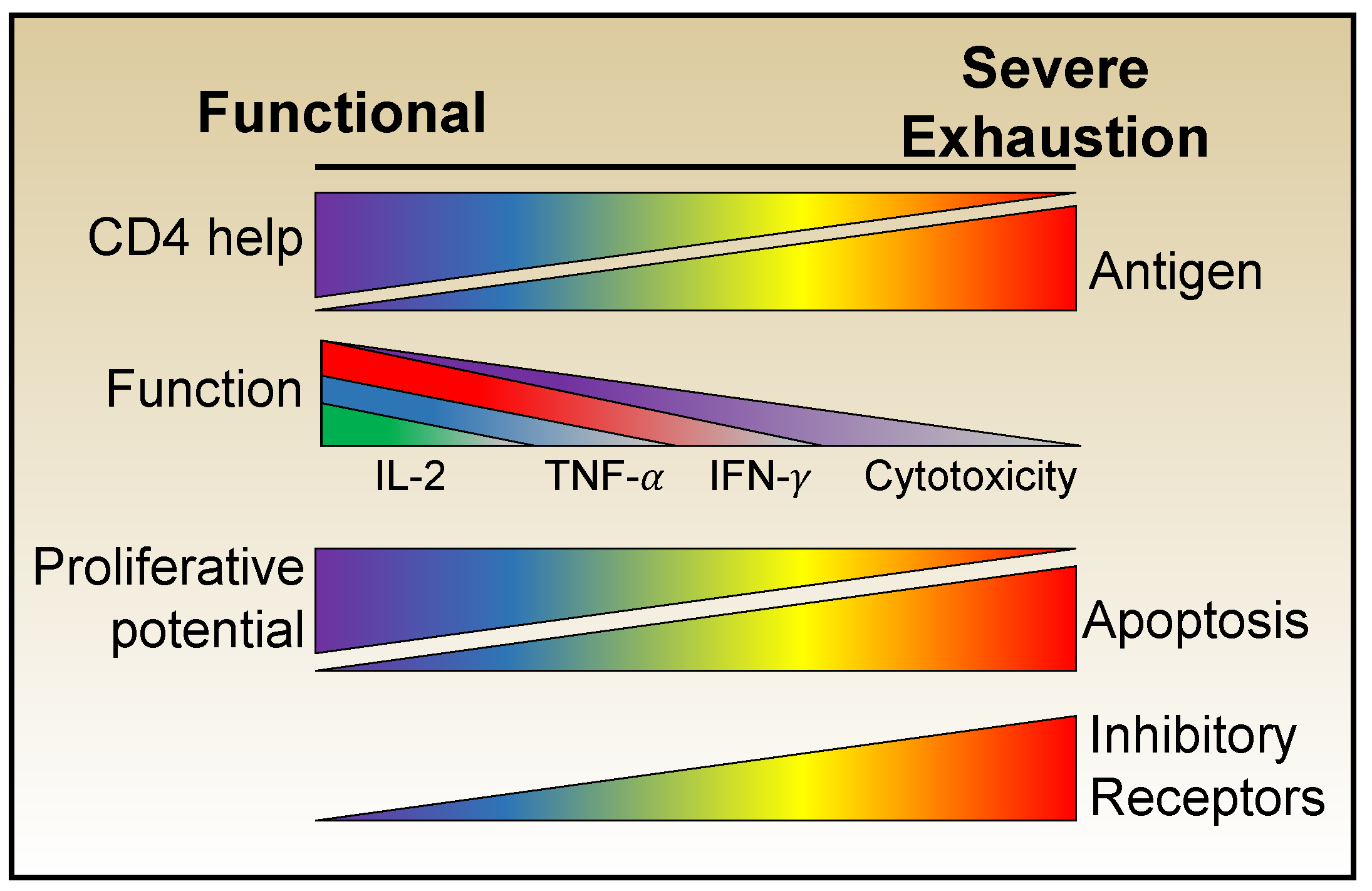
2.1. Loss of Function
2.2. Inhibitory Receptors
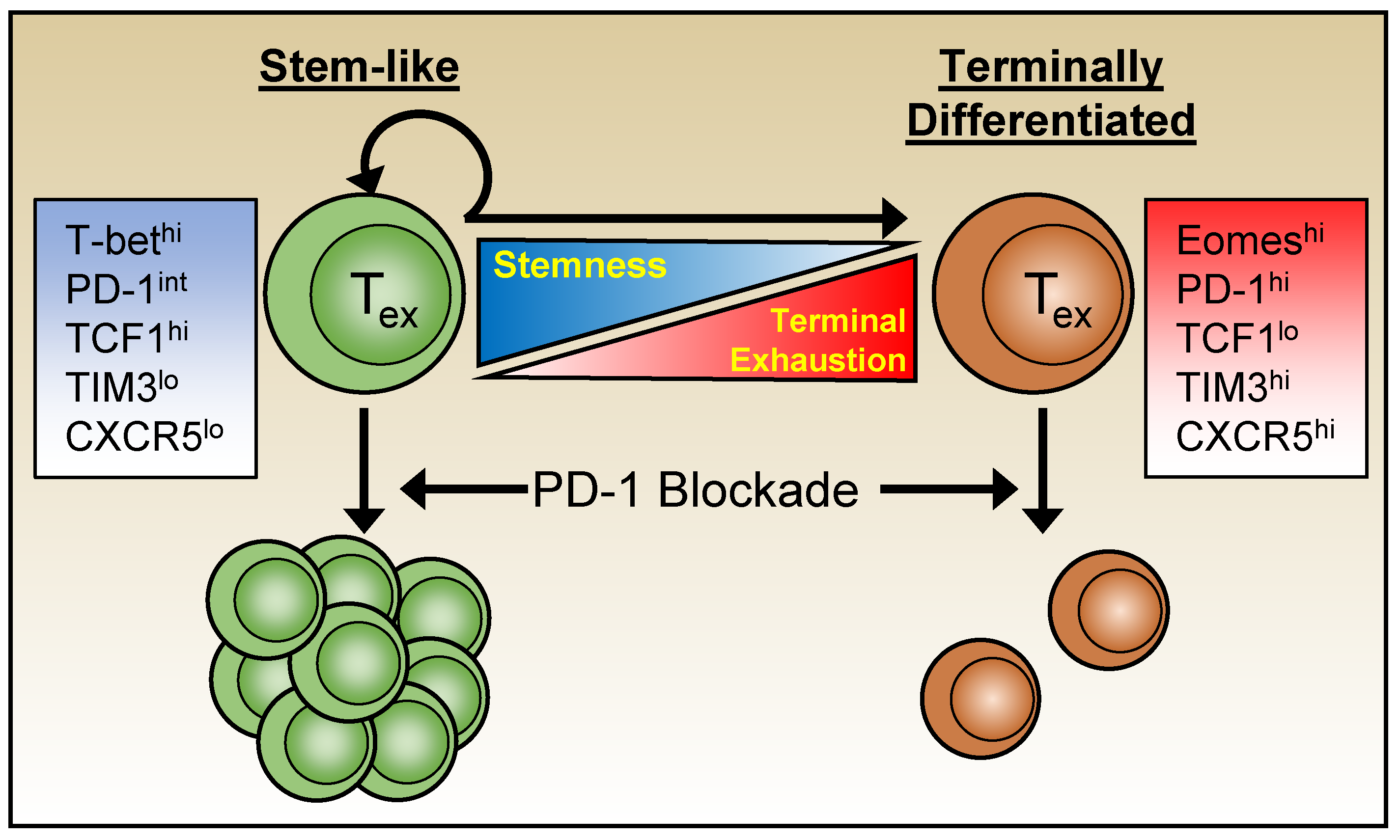
2.3. Transcriptional Regulators
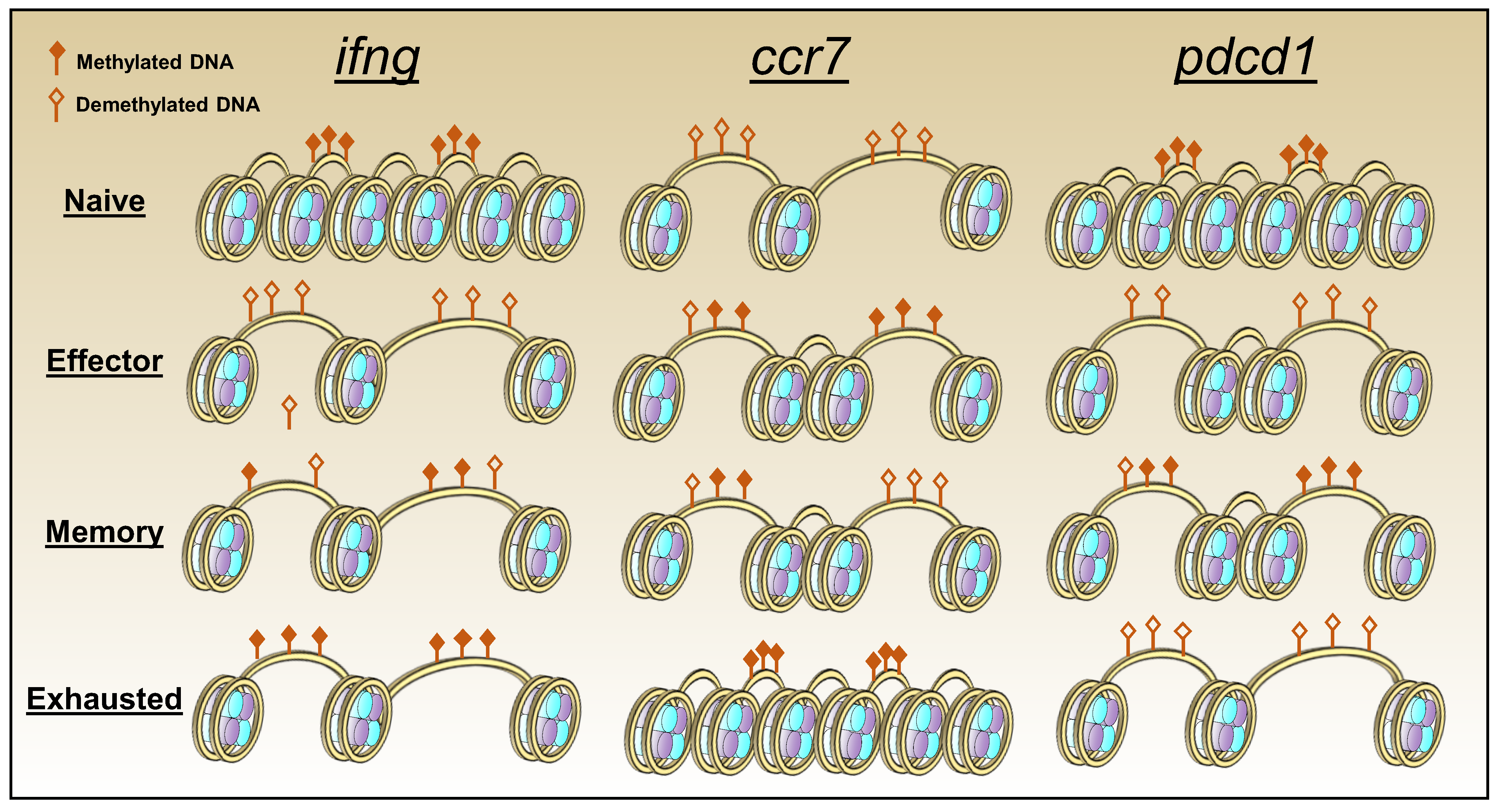
2.4. Epigenetics
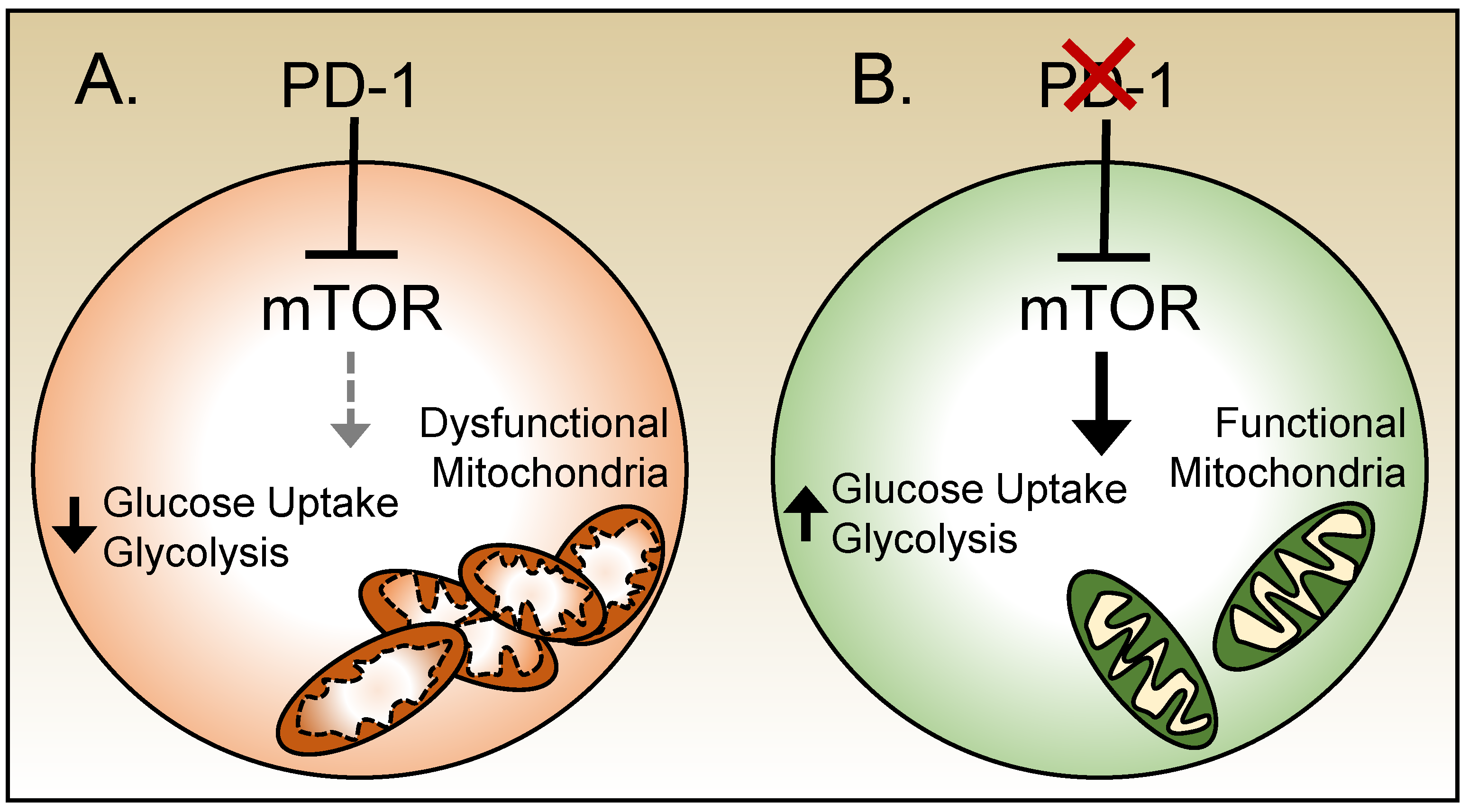
2.5. Metabolism
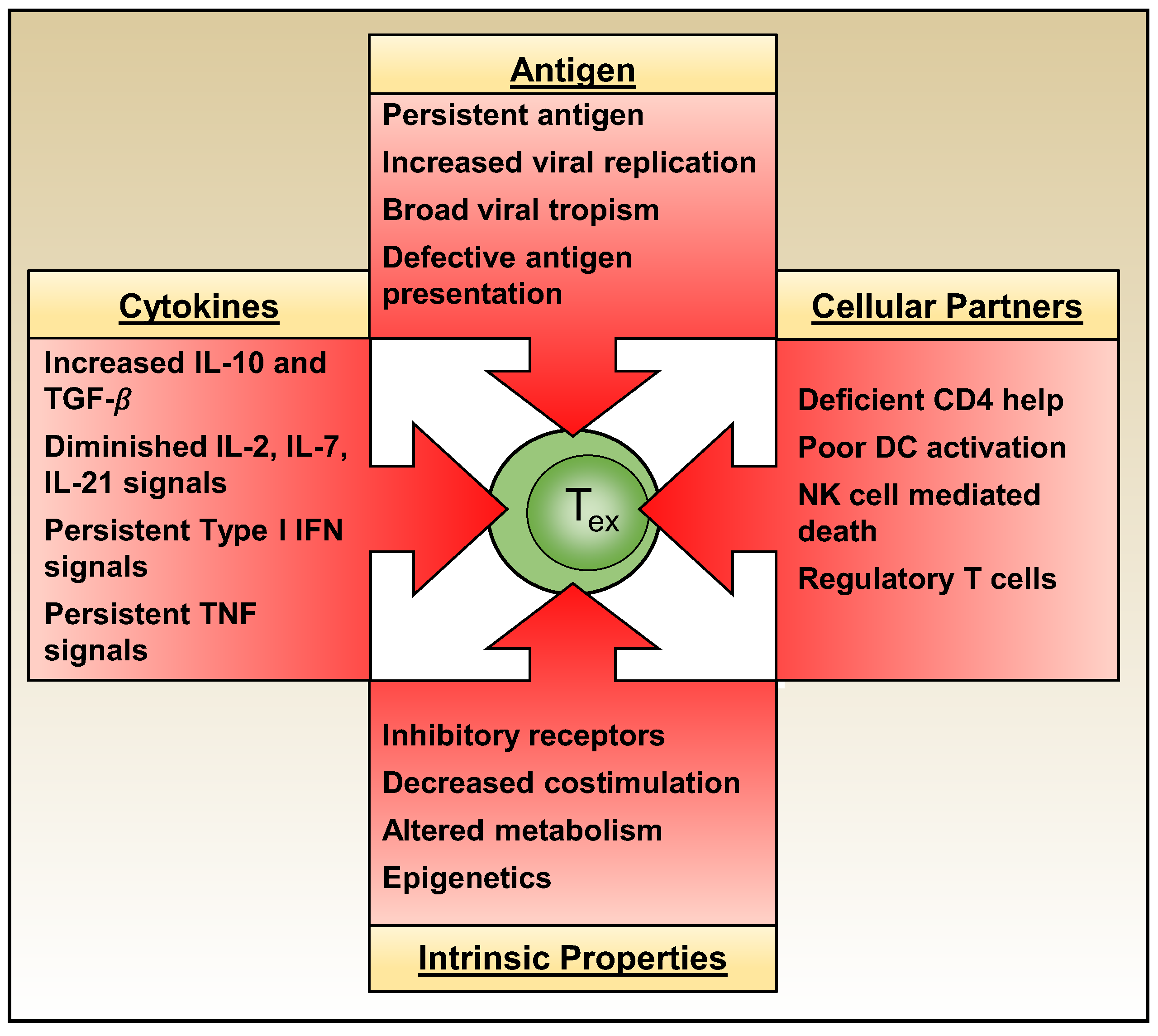
3. Extrinsic Drivers of T Cell Exhaustion
3.1. Antigenic Signals
3.2. Cellular Partners
3.2.1. Ineffective CD4 T Cell Help
3.2.2. Natural Killer (NK) Cells
3.3. Cytokines
3.3.1. Proinflammatory Cytokines
3.3.2. Immunosuppression by IL-10 and TGF-β
3.3.3. Common-Gamma Chain Receptor Family Cytokines: IL-2, IL-7, IL-15, and IL-21
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armstrong, C.; Lillie, R.D. Experimental Lymphocytic Choriomeningitis of Monkeys and Mice Produced by a Virus Encountered in Studies of the 1933 St. Louis Encephalitis Epidemic. Public Health Rep. 1934, 49, 1019–1027. [Google Scholar] [CrossRef]
- Traub, E. A Filterable Virus Recovered from White Mice. Science 1935, 81, 298–299. [Google Scholar] [CrossRef]
- Raju, T.N. The Nobel Chronicles. Lancet 2000, 356, 172. [Google Scholar] [CrossRef]
- Raju, T.N. The Nobel Chronicles. Lancet 1999, 353, 2253. [Google Scholar] [CrossRef]
- Ahmed, R.; Salmi, A.; Butler, L.D.; Chiller, J.M.; Oldstone, M.B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 1984, 160, 521–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergthaler, A.; Flatz, L.; Hegazy, A.N.; Johnson, S.; Horvath, E.; Löhning, M.; Pinschewer, D.D. Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 21641–21646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrow, P.; Evans, C.F.; Oldstone, M.B. Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J. Virol. 1995, 69, 1059–1070. [Google Scholar] [PubMed]
- Mueller, S.N.; Matloubian, M.; Clemens, D.M.; Sharpe, A.H.; Freeman, G.J.; Gangappa, S.; Larsen, C.P.; Ahmed, R. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. USA 2007, 104, 15430–15435. [Google Scholar] [CrossRef] [Green Version]
- Salvato, M.; Borrow, P.; Shimomaye, E.; Oldstone, M.B. Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J. Virol. 1991, 65, 1863–1869. [Google Scholar]
- Sevilla, N.; Kunz, S.; Holz, A.; Lewicki, H.; Homann, D.; Yamada, H.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B.A. Immunosuppression and Resultant Viral Persistence by Specific Viral Targeting of Dendritic Cells. J. Exp. Med. 2000, 192, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, B.M.; Emonet, S.F.; Welch, M.J.; Lee, A.M.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc. Natl. Acad. Sci. USA 2011, 108, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, R.; Oldstone, M.B. Organ-specific selection of viral variants during chronic infection. J. Exp. Med. 1988, 167, 1719–1724. [Google Scholar] [CrossRef]
- Matloubian, M.; Somasundaram, T.; Kolhekar, S.R.; Selvakumar, R.; Ahmed, R. Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. J. Exp. Med. 1990, 172, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Sevilla, N.; McGavern, D.B.; Teng, C.; Kunz, S.; Oldstone, M.B.A. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J. Clin. Investig. 2004, 113, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, B.M.; Teijaro, J.R.; de la Torre, J.C.; Oldstone, M.B.A. Early Virus-Host Interactions Dictate the Course of a Persistent Infection. PLoS Pathog. 2015, 11, e1004588. [Google Scholar] [CrossRef]
- Baca Jones, C.; Filippi, C.; Sachithanantham, S.; Rodriguez-Calvo, T.; Ehrhardt, K.; von Herrath, M. Direct Infection of Dendritic Cells during Chronic Viral Infection Suppresses Antiviral T Cell Proliferation and Induces IL-10 Expression in CD4 T Cells. PLoS ONE 2014, 9, e90855. [Google Scholar] [CrossRef]
- Ng, C.T.; Oldstone, M.B.A. Infected CD8α− dendritic cells are the predominant source of IL-10 during establishment of persistent viral infection. Proc. Natl. Acad. Sci. USA 2012, 109, 14116–14121. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.I.; Liou, L.-Y.; Mack, L.; Mendoza, M.; Oldstone, M.B.A. Persistent Virus Infection Inhibits Type I Interferon Production by Plasmacytoid Dendritic Cells to Facilitate Opportunistic Infections. Cell Host Microbe 2008, 4, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.N.; Germain, R.N. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat. Rev. Immunol. 2009, 9, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.F.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B.A. Persistent LCMV Infection Is Controlled by Blockade of Type I Interferon Signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte, M.A.; Beliën, J.A.M.; Schadee-Eestermans, I.; Jansen, W.; Unger, W.W.J.; van Rooijen, N.; Kraal, G.; Mebius, R.E. A Conduit System Distributes Chemokines and Small Blood-borne Molecules through the Splenic White Pulp. J. Exp. Med. 2003, 198, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanjappa, S.G.; Kim, E.H.; Suresh, M. Immunotherapeutic effects of IL-7 during a chronic viral infection in mice. Blood 2011, 117, 5123–5132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, M.; Calzascia, T.; Toe, J.G.; Preston, S.P.; Lin, A.E.; Elford, A.R.; Shahinian, A.; Lang, P.A.; Lang, K.S.; Morre, M.; et al. IL-7 Engages Multiple Mechanisms to Overcome Chronic Viral Infection and Limit Organ Pathology. Cell 2011, 144, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, Y.; Moysi, E.; Petrovas, C. The Lymph Node in HIV Pathogenesis. Curr. HIV/AIDS Rep. 2017, 14, 133–140. [Google Scholar] [CrossRef]
- Zeng, M.; Southern, P.J.; Reilly, C.S.; Beilman, G.J.; Chipman, J.G.; Schacker, T.W.; Haase, A.T. Lymphoid Tissue Damage in HIV-1 Infection Depletes Naïve T Cells and Limits T Cell Reconstitution after Antiretroviral Therapy. PLOS Pathog. 2012, 8, e1002437. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, A.; Glithero, A.; Godkin, A.; Tissot, A.C.; Plückthun, A.; Elliott, T.; Hengartner, H.; Zinkernagel, R. Induction and Exhaustion of Lymphocytic Choriomeningitis Virus–specific Cytotoxic T Lymphocytes Visualized Using Soluble Tetrameric Major Histocompatibility Complex Class I–Peptide Complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.D.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral Immune Evasion Due to Persistence of Activated T Cells Without Effector Function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [Green Version]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of Hepatitis B Virus (HBV)-Specific T-Cell Dysfunction in Chronic HBV Infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [Green Version]
- Day, C.L.; Abrahams, D.A.; Lerumo, L.; van Rensburg, E.J.; Stone, L.; O’rie, T.; Pienaar, B.; de Kock, M.; Kaplan, G.; Mahomed, H.; et al. Functional Capacity of Mycobacterium tuberculosis-Specific T Cell Responses in Humans Is Associated with Mycobacterial Load. J. Immunol. 2011, 187, 2222–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Far, M.; Halwani, R.; Said, E.; Trautmann, L.; Doroudchi, M.; Janbazian, L.; Fonseca, S.; van Grevenynghe, J.; Yassine-Diab, B.; Sékaly, R.-P.; et al. T-cell exhaustion in HIV infection. Curr. HIV/AIDS Rep. 2008, 5, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Radziewicz, H.; Ibegbu, C.C.; Fernandez, M.L.; Workowski, K.A.; Obideen, K.; Wehbi, M.; Hanson, H.L.; Steinberg, J.P.; Masopust, D.; Wherry, E.J.; et al. Liver-Infiltrating Lymphocytes in Chronic Human Hepatitis C Virus Infection Display an Exhausted Phenotype with High Levels of PD-1 and Low Levels of CD127 Expression. J. Virol. 2007, 81, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.N.; Horne-Debets, J.M.; Leow, C.-Y.; Karunarathne, D.S. Malaria drives T cells to exhaustion. Front. Microbiol. 2014, 5, 249. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [Green Version]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.J.; Khanolkar, A.; Tebo, A.E.; Zajac, A.J. Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J. Immunol. 2004, 172, 4204–4214. [Google Scholar] [CrossRef]
- Fuller, M.J.; Zajac, A.J. Ablation of CD8 and CD4 T cell responses by high viral loads. J. Immunol. 2003, 170, 477–486. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [Green Version]
- Agnellini, P.; Wolint, P.; Rehr, M.; Cahenzli, J.; Karrer, U.; Oxenius, A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc. Natl. Acad. Sci. USA 2007, 104, 4565–4570. [Google Scholar] [CrossRef] [Green Version]
- Graw, F.; Richter, K.; Oxenius, A.; Regoes, R.R. Comparison of cytotoxic T lymphocyte efficacy in acute and persistent lymphocytic choriomeningitis virus infection. Proc. R. Soc. B 2011, 278, 3395–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Bauer, D.E.; Tuttleton, S.E.; Lewin, S.; Gettie, A.; Blanchard, J.; Irwin, C.E.; Safrit, J.T.; Mittler, J.; Weinberger, L.; et al. Dramatic Rise in Plasma Viremia after CD8+ T Cell Depletion in Simian Immunodeficiency Virus–infected Macaques. J. Exp. Med. 1999, 189, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.E.; Kuroda, M.J.; Santra, S.; Sasseville, V.G.; Simon, M.A.; Lifton, M.A.; Racz, P.; Tenner-Racz, K.; Dalesandro, M.; Scallon, B.J.; et al. Control of Viremia in Simian Immunodeficiency Virus Infection by CD8+ Lymphocytes. Science 1999, 283, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.T.; Yi, J.S.; Zajac, A.J. Exhausted CD8 T cells downregulate the IL-18 receptor and become unresponsive to inflammatory cytokines and bacterial co-infections. PLoS Pathog. 2011, 7, e1002273. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2008, 10, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Jin, H.-T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.-J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.J.; Yu, X.; Grogan, J.L. The checkpoint inhibitor TIGIT limits antitumor and antiviral CD8+ T cell responses. Oncoimmunology 2015, 4, e1036214. [Google Scholar] [CrossRef]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Agnellini, P.; Oxenius, A. On the role of the inhibitory receptor LAG-3 in acute and chronic LCMV infection. Int. Immunol. 2010, 22, 13–23. [Google Scholar] [CrossRef]
- Odorizzi, P.M.; Wherry, E.J. Inhibitory Receptors on Lymphocytes: Insights from Infections. J. Immunol. 2012, 188, 2957–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, C.W.; Zajac, A.J.; Jamieson, A.M.; Corral, L.; Hammer, G.E.; Ahmed, R.; Raulet, D.H. Viral and Bacterial Infections Induce Expression of Multiple NK Cell Receptors in Responding CD8+ T Cells. J. Immunol. 2002, 169, 1444–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, S.D.; Crawford, A.; Shin, H.; Polley, A.; Freeman, G.J.; Wherry, E.J. Tissue-Specific Differences in PD-1 and PD-L1 Expression during Chronic Viral Infection: Implications for CD8 T-Cell Exhaustion. J. Virol. 2010, 84, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Blattman, J.N.; Wherry, E.J.; Ha, S.-J.; van der Most, R.G.; Ahmed, R. Impact of Epitope Escape on PD-1 Expression and CD8 T-Cell Exhaustion during Chronic Infection. J. Virol. 2009, 83, 4386–4394. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Petrovas, C.; Price, D.A.; Mattapallil, J.; Ambrozak, D.R.; Geldmacher, C.; Cecchinato, V.; Vaccari, M.; Tryniszewska, E.; Gostick, E.; Roederer, M.; et al. SIV-specific CD8+ T cells express high levels of PD1 and cytokines but have impaired proliferative capacity in acute and chronic SIVmac251 infection. Blood 2007, 110, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbani, S.; Amadei, B.; Tola, D.; Massari, M.; Schivazappa, S.; Missale, G.; Ferrari, C. PD-1 Expression in Acute Hepatitis C Virus (HCV) Infection Is Associated with HCV-Specific CD8 Exhaustion. J. Virol. 2006, 80, 11398–11403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afanasiev, O.K.; Yelistratova, L.; Miller, N.; Nagase, K.; Paulson, K.; Iyer, J.G.; Ibrani, D.; Koelle, D.M.; Nghiem, P. Merkel Polyomavirus-Specific T Cells Fluctuate with Merkel Cell Carcinoma Burden and Express Therapeutically Targetable PD-1 and Tim-3 Exhaustion Markers. Clin. Cancer Res. 2013, 19, 5351–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwacz, K.; Bricogne, C.; MacDonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 2011, 3, 581–592. [Google Scholar] [CrossRef]
- Shamim, M.; Nanjappa, S.G.; Singh, A.; Plisch, E.H.; LeBlanc, S.E.; Walent, J.; Svaren, J.; Seroogy, C.; Suresh, M. Cbl-b Regulates Antigen-Induced TCR Down-Regulation and IFN-γ Production by Effector CD8 T Cells without Affecting Functional Avidity. J. Immunol. 2007, 179, 7233–7243. [Google Scholar] [CrossRef]
- Wei, F.; Zhong, S.; Ma, Z.; Kong, H.; Medvec, A.; Ahmed, R.; Freeman, G.J.; Krogsgaard, M.; Riley, J.L. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. USA 2013, 110, E2480–E2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8+ T Cells during Chronic Infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [Green Version]
- Zinselmeyer, B.H.; Heydari, S.; Sacristán, C.; Nayak, D.; Cammer, M.; Herz, J.; Cheng, X.; Davis, S.J.; Dustin, M.L.; McGavern, D.B. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J. Exp. Med. 2013, 210, 757–774. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1200694?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dwww.ncbi.nlm.nih.gov (accessed on 14 December 2018).
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti–PD-1 Therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.-R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Avery, L.; Filderman, J.; Szymczak-Workman, A.L.; Kane, L.P. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. USA 2018, 115, 2455–2460. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Kamphorst, A.O.; Wieland, A.; Araki, K.; Iyer, S.S.; West, E.E.; O’Mara, L.; Yang, S.; Konieczny, B.T.; Sharpe, A.H.; et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J. Exp. Med. 2014, 211, 1905–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, E.E.; Jin, H.-T.; Rasheed, A.-U.; Penaloza-MacMaster, P.; Ha, S.-J.; Tan, W.G.; Youngblood, B.; Freeman, G.J.; Smith, K.A.; Ahmed, R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Investig. 2013, 123, 2604–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Morita, M.; Bederman, A.G.; Konieczny, B.T.; Kissick, H.T.; Sonenberg, N.; Ahmed, R. Translation is actively regulated during the differentiation of CD8+ effector T cells. Nat. Immunol. 2017, 18, 1046–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network Analysis Reveals Centrally Connected Genes and Pathways Involved in CD8+ T Cell Exhaustion versus Memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef]
- Mackerness, K.J.; Cox, M.A.; Lilly, L.M.; Weaver, C.T.; Harrington, L.E.; Zajac, A.J. Pronounced virus-dependent activation drives exhaustion but sustains IFN-γ transcript levels. J. Immunol. 2010, 185, 3643–3651. [Google Scholar] [CrossRef] [PubMed]
- Salerno, F.; Engels, S.; van den Biggelaar, M.; van Alphen, F.P.J.; Guislain, A.; Zhao, W.; Hodge, D.L.; Bell, S.E.; Medema, J.P.; von Lindern, M.; et al. Translational repression of pre-formed cytokine-encoding mRNA prevents chronic activation of memory T cells. Nat. Immunol. 2018, 19, 828. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grusdat, M.; McIlwain, D.R.; Xu, H.C.; Pozdeev, V.I.; Knievel, J.; Crome, S.Q.; Robert-Tissot, C.; Dress, R.J.; Pandyra, A.A.; Speiser, D.E.; et al. IRF4 and BATF are critical for CD8+ T-cell function following infection with LCMV. Cell Death Differ. 2014, 21, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Leong, Y.A.; Chen, Y.; Ong, H.S.; Wu, D.; Man, K.; Deleage, C.; Minnich, M.; Meckiff, B.J.; Wei, Y.; Hou, Z.; et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 2016, 17, 1187–1196. [Google Scholar] [CrossRef]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129.e5–1141.e5. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef]
- Wu, T.; Ji, Y.; Moseman, E.A.; Xu, H.C.; Manglani, M.; Kirby, M.; Anderson, S.M.; Handon, R.; Kenyon, E.; Elkahloun, A.; et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Xin, G.; Schauder, D.M.; Lainez, B.; Weinstein, J.S.; Dai, Z.; Chen, Y.; Esplugues, E.; Wen, R.; Wang, D.; Parish, I.A.; et al. A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Rep. 2015, 13, 1118–1124. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Youngblood, B.A.; Austin, J.W.; Mohammed, A.U.R.; Butler, R.; Ahmed, R.; Boss, J.M. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 2014, 211, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 Regulates PD-1 Expression upon T Cell Activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A Role for the Transcriptional Repressor Blimp-1 in CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, C.; Oestreich, K.J.; Paley, M.A.; Crawford, A.; Angelosanto, J.M.; Ali, M.-A.A.; Intlekofer, A.M.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Hou, S.; Liu, C.; Zhang, A.; Bai, Q.; Han, M.; Yang, Y.; Wei, G.; Shen, T.; Yang, X.; et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 2016, 537, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Scott-Browne, J.P.; López-Moyado, I.F.; Trifari, S.; Wong, V.; Chavez, L.; Rao, A.; Pereira, R.M. Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity 2016, 45, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelosanto, J.M.; Blackburn, S.D.; Crawford, A.; Wherry, E.J. Progressive Loss of Memory T Cell Potential and Commitment to Exhaustion during Chronic Viral Infection. J. Virol. 2012, 86, 8161–8170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utzschneider, D.T.; Legat, A.; Fuertes Marraco, S.A.; Carrié, L.; Luescher, I.; Speiser, D.E.; Zehn, D. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat. Immunol. 2013, 14, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhou, X.; DiSpirito, J.R.; Wang, C.; Wang, Y.; Shen, H. Epigenetic Manipulation Restores Functions of Defective CD8+ T Cells From Chronic Viral Infection. Mol. Ther. 2014, 22, 1698–1706. [Google Scholar] [CrossRef]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine Metabolism Regulates Maintenance and Differentiation of Human Pluripotent Stem Cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Pearce, E.L. Emerging concepts of T Cell Metab. as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T Cell Memory by Modulating Fatty Acid Metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metab. and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Barber, D.L.; Kaech, S.M.; Blattman, J.N.; Ahmed, R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 16004–16009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornberg, M.; Kenney, L.L.; Chen, A.T.; Waggoner, S.N.; Kim, S.-K.; Dienes, H.P.; Welsh, R.M.; Selin, L.K. Clonal exhaustion as a mechanism to protect against severe immunopathology and death from an overwhelming CD8 T cell response. Front. Immunol. 2013, 4, 475. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Battegay, M.; van den Broek, M.; Laine, E.; Hoffmann-Rohrer, U.; Zinkernagel, R.M. Role of virus and host variables in virus persistence or immunopathological disease caused by a non-cytolytic virus. J. Gen. Virol. 1995, 76, 381–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, M.R. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 2006, 107, 4781–4789. [Google Scholar] [CrossRef] [PubMed]
- Oxenius, A.; Sewell, A.K.; Dawson, S.J.; Günthard, H.F.; Fischer, M.; Gillespie, G.M.; Rowland-Jones, S.L.; Fagard, C.; Hirschel, B.; Phillips, R.E.; et al. Functional Discrepancies in HIV-Specific CD8<sup>+</sup> T-Lymphocyte Populations Are Related to Plasma Virus Load. J. Clin. Immunol. 2002, 22, 363–374. [Google Scholar]
- Moskophidis, D.; Zinkernagel, R.M. Immunobiology of cytotoxic T-cell escape mutants of lymphocytic choriomeningitis virus. J. Virol. 1995, 69, 2187–2193. [Google Scholar]
- Butz, E.A.; Bevan, M.J. Differential Presentation of the Same MHC Class I Epitopes by Fibroblasts and Dendritic Cells. J. Immunol. 1998, 160, 2139–2144. [Google Scholar]
- Utzschneider, D.T.; Alfei, F.; Roelli, P.; Barras, D.; Chennupati, V.; Darbre, S.; Delorenzi, M.; Pinschewer, D.D.; Zehn, D. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J. Exp. Med. 2016, 213, 1819–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Brocker, T.; Oxenius, A. Antigen amount dictates CD8+T-cell exhaustion during chronic viral infection irrespective of the type of antigen presenting cell. Eur. J. Immunol. 2012, 42, 2290–2304. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Blackburn, S.D.; Blattman, J.N.; Wherry, E.J. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 2007, 204, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Battegay, M.; Moskophidis, D.; Rahemtulla, A.; Hengartner, H.; Mak, T.W.; Zinkernagel, R.M. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J. Virol. 1994, 68, 4700–4704. [Google Scholar]
- Matloubian, M.; Concepcion, R.J.; Ahmed, R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 1994, 68, 8056–8063. [Google Scholar] [PubMed]
- Wiesel, M.; Oxenius, A. From crucial to negligible: functional CD8+ T-cell responses and their dependence on CD4+ T-cell help. Eur. J. Immunol. 2012, 42, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, L.; Klenerman, P.; Zinkernagel, R.M.; Ehl, S. Exhaustion of cytotoxic T cells during adoptive immunotherapy of virus carrier mice can be prevented by B cells or CD4+ T cells. Eur. J. Immunol. 2002, 32, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Aubert, R.D.; Kamphorst, A.O.; Sarkar, S.; Vezys, V.; Ha, S.-J.; Barber, D.L.; Ye, L.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc. Natl. Acad. Sci. USA 2011, 108, 21182–21187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, S.R.M.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.A.P.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.; Huang, A.Y.; Altan-Bonnet, G.; Stoll, S.; Scheinecker, C.; Germain, R.N. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell–dendritic cell interaction. Nature 2006, 440, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef]
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.E.M.; van der Voort, E.I.H.; Offringa, R.; Melief, C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Wiesel, M.; Joller, N.; Ehlert, A.-K.; Crouse, J.; Spörri, R.; Bachmann, M.F.; Oxenius, A. Th Cells Act Via Two Synergistic Pathways To Promote Antiviral CD8+ T Cell Responses. J. Immunol. 2010, 185, 5188–5197. [Google Scholar] [CrossRef]
- Brooks, D.G.; Teyton, L.; Oldstone, M.B.A.; McGavern, D.B. Intrinsic Functional Dysregulation of CD4 T Cells Occurs Rapidly following Persistent Viral Infection. J. Virol. 2005, 79, 10514–10527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, A.; Angelosanto, J.M.; Kao, C.; Doering, T.A.; Odorizzi, P.M.; Barnett, B.E.; Wherry, E.J. Molecular and Transcriptional Basis of CD4+ T Cell Dysfunction during Chronic Infection. Immunity 2014, 40, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxenius, A.; Zinkernagel, R.M.; Hengartner, H. Comparison of Activation versus Induction of Unresponsiveness of Virus-Specific CD4+ and CD8+ T Cells upon Acute versus Persistent Viral Infection. Immunity 1998, 9, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Fahey, L.M.; Wilson, E.B.; Elsaesser, H.; Fistonich, C.D.; McGavern, D.B.; Brooks, D.G. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J. Exp. Med. 2011, 208, 987–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osokine, I.; Snell, L.M.; Cunningham, C.R.; Yamada, D.H.; Wilson, E.B.; Elsaesser, H.J.; de la Torre, J.C.; Brooks, D. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. USA 2014, 111, 7409–7414. [Google Scholar] [CrossRef] [Green Version]
- Greczmiel, U.; Krautler, N.J.; Pedrioli, A.; Bartsch, I.; Agnellini, P.; Bedenikovic, G.; Harker, J.A.; Richter, K.; Oxenius, A. Sustained T follicular helper cell response is essential for control of chronic LCMV infection. Sci. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Harker, J.A.; Lewis, G.M.; Mack, L.; Zuniga, E.I. Late Interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 2011, 334, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Punkosdy, G.A.; Blain, M.; Glass, D.D.; Lozano, M.M.; O’Mara, L.; Dudley, J.P.; Ahmed, R.; Shevach, E.M. Regulatory T-cell expansion during chronic viral infection is dependent on endogenous retroviral superantigens. Proc. Natl. Acad. Sci. USA 2011, 108, 3677–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, I.; Schneider, C.; Fröhlich, A.; Frebel, H.; Christ, D.; Leonard, W.J.; Sparwasser, T.; Oxenius, A.; Freigang, S.; Kopf, M. IL-21 Restricts Virus-driven Treg Cell Expansion in Chronic LCMV Infection. PLoS Pathog. 2013, 9, e1003362. [Google Scholar] [CrossRef]
- Veiga-Parga, T.; Sehrawat, S.; Rouse, B.T. Role of regulatory T cells during virus infection. Immunol. Rev. 2013, 255, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Crome, S.Q.; Lang, P.A.; Lang, K.S.; Ohashi, P.S. Natural killer cells regulate diverse T cell responses. Trends Immunol. 2013, 34, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.D.; Whitmire, J.K. The Depletion of NK Cells Prevents T Cell Exhaustion to Efficiently Control Disseminating Virus Infection. J. Immunol. 2013, 190, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Lang, K.S.; Xu, H.C.; Grusdat, M.; Parish, I.A.; Recher, M.; Elford, A.R.; Dhanji, S.; Shaabani, N.; Tran, C.W.; et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, S.N.; Cornberg, M.; Selin, L.K.; Welsh, R.M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2012, 481, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, S.N.; Daniels, K.A.; Welsh, R.M. Therapeutic Depletion of Natural Killer Cells Controls Persistent Infection. J. Virol. 2014, 88, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Bedenikovic, G.; Wiesel, M.; Ibberson, M.; Xenarios, I.; Von Laer, D.; Kalinke, U.; Vivier, E.; Jonjic, S.; Oxenius, A. Type I Interferons Protect T Cells against NK Cell Attack Mediated by the Activating Receptor NCR1. Immunity 2014, 40, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.C.; Grusdat, M.; Pandyra, A.A.; Polz, R.; Huang, J.; Sharma, P.; Deenen, R.; Köhrer, K.; Rahbar, R.; Diefenbach, A.; et al. Type I Interferon Protects Antiviral CD8+ T Cells from NK Cell Cytotoxicity. Immunity 2014, 40, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Jacquelin, B.; Mayau, V.; Targat, B.; Liovat, A.-S.; Kunkel, D.; Petitjean, G.; Dillies, M.-A.; Roques, P.; Butor, C.; Silvestri, G.; et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Investig. 2009, 119, 3544–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of Chronic Type I Interferon Signaling to Control Persistent LCMV Infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, A.; Rezek, V.; Youn, C.; Lam, B.; Chang, N.; Rick, J.; Carrillo, M.; Martin, H.; Kasparian, S.; Syed, P.; et al. Targeting type I interferon–mediated activation restores immune function in chronic HIV infection. J. Clin. Investig. 2017, 127, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Sullivan, B.M.; Teijaro, J.R.; Lee, A.M.; Welch, M.; Rice, S.; Sheehan, K.C.F.; Schreiber, R.D.; Oldstone, M.B.A. Blockade of Interferon Beta, but Not Interferon Alpha, Signaling Controls Persistent Viral Infection. Cell Host Microbe 2015, 17, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, C.R.; Champhekar, A.; Tullius, M.V.; Dillon, B.J.; Zhen, A.; de la Fuente, J.R.; Herskovitz, J.; Elsaesser, H.; Snell, L.M.; Wilson, E.B.; et al. Type I and Type II Interferon Coordinately Regulate Suppressive Dendritic Cell Fate and Function during Viral Persistence. PLOS Pathog. 2016, 12, e1005356. [Google Scholar] [CrossRef] [PubMed]
- Maine, C.J.; Teijaro, J.R.; Marquardt, K.; Sherman, L.A. PTPN22 contributes to exhaustion of T lymphocytes during chronic viral infection. Proc. Natl. Acad. Sci. USA 2016, 113, E7231–E7239. [Google Scholar] [CrossRef] [PubMed]
- Otero, D.C.; Fares-Frederickson, N.J.; Xiao, M.; Baker, D.P.; David, M. IFN-β Selectively Inhibits IL-2 Production through CREM-Mediated Chromatin Remodeling. J. Immunol 2015, 194, 5120–5128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Swiecki, M.; Cella, M.; Alber, G.; Schreiber, R.D.; Gilfillan, S.; Colonna, M. Timing and Magnitude of Type I Interferon Responses by Distinct Sensors Impact CD8 T Cell Exhaustion and Chronic Viral Infection. Cell Host Microbe 2012, 11, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harker, J.A.; Dolgoter, A.; Zuniga, E.I. Cell-Intrinsic IL-27 and gp130 Cytokine Receptor Signaling Regulates Virus-Specific CD4+ T Cell Responses and Viral Control during Chronic Infection. Immunity 2013, 39, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, M.; Abdullah, Z.; Chemnitz, J.M.; Maisel, D.; Sander, J.; Lehmann, C.; Thabet, Y.; Shinde, P.V.; Schmidleithner, L.; Köhne, M.; et al. Tumor-necrosis factor impairs CD4+ T cell–mediated immunological control in chronic viral infection. Nat. Immunol. 2016, 17, 593–603. [Google Scholar] [CrossRef]
- Wilson, E.B.; Brooks, D.G. The Role of IL-10 in Regulating Immunity to Persistent Viral Infections. In Negative Co-Receptors and Ligands; Current Topics in Microbiology and Immunology; Ahmed, R., Honjo, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 39–65. ISBN 978-3-642-19544-0. [Google Scholar]
- Brockman, M.A.; Kwon, D.S.; Tighe, D.P.; Pavlik, D.F.; Rosato, P.C.; Sela, J.; Porichis, F.; Le Gall, S.; Waring, M.T.; Moss, K.; et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 2009, 114, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B.A. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Clerici, M.; Wynn, T.A.; Berzofsky, J.A.; Blatt, S.P.; Hendrix, C.W.; Sher, A.; Coffman, R.L.; Shearer, G.M. Role of interleukin-10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J. Clin. Investig. 1994, 93, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyodo, N.; Nakamura, I.; Imawari, M. Hepatitis B core antigen stimulates interleukin-10 secretion by both T cells and monocytes from peripheral blood of patients with chronic hepatitis B virus infection. Clin. Exp. Immunol. 2004, 135, 462–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, D.E.; Ikeda, F.; Li, Y.; Nakamoto, N.; Ganesan, S.; Valiga, M.E.; Nunes, F.A.; Rajender Reddy, K.; Chang, K.-M. Peripheral virus-specific T-cell interleukin-10 responses develop early in acute hepatitis C infection and become dominant in chronic hepatitis. J. Hepatol. 2008, 48, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, C.H.; Chappell, C.P.; Jacob, J. Interleukin-10 plays an early role in generating virus-specific T cell anergy. BMC Immunol. 2007, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Ohga, S.; Nomura, A.; Takada, H.; Tanaka, T.; Furuno, K.; Takahata, Y.; Kinukawa, N.; Fukushima, N.; Imai, S.; Hara, T. Dominant expression of interleukin-10 and transforming growth factor-β genes in activated T-cells of chronic active epstein–barr virus infection. J. Med. Virol. 2004, 74, 449–458. [Google Scholar] [CrossRef]
- Richter, K.; Perriard, G.; Behrendt, R.; Schwendener, R.A.; Sexl, V.; Dunn, R.; Kamanaka, M.; Flavell, R.A.; Roers, A.; Oxenius, A. Macrophage and T Cell Produced IL-10 Promotes Viral Chronicity. PLoS Pathog. 2013, 9, e1003735. [Google Scholar] [CrossRef]
- Smith, L.K.; Boukhaled, G.M.; Condotta, S.A.; Mazouz, S.; Guthmiller, J.J.; Vijay, R.; Butler, N.S.; Bruneau, J.; Shoukry, N.H.; Krawczyk, C.M.; et al. Interleukin-10 Directly Inhibits CD8+ T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 2018, 48, 299–312. [Google Scholar] [CrossRef]
- Richter, K.; Perriard, G.; Oxenius, A. Reversal of chronic to resolved infection by IL-10 blockade is LCMV strain dependent. Eur. J. Immunol. 2013, 43, 649–654. [Google Scholar] [CrossRef] [Green Version]
- Brooks, D.G.; Ha, S.-J.; Elsaesser, H.; Sharpe, A.H.; Freeman, G.J.; Oldstone, M.B.A. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc. Natl. Acad. Sci. USA 2008, 105, 20428–20433. [Google Scholar] [CrossRef] [Green Version]
- Xin, G.; Zander, R.; Schauder, D.M.; Chen, Y.; Weinstein, J.S.; Drobyski, W.R.; Tarakanova, V.; Craft, J.; Cui, W. Single-cell RNA sequencing unveils an IL-10-producing helper subset that sustains humoral immunity during persistent infection. Nat. Commun. 2018, 9, 5037. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming Growth Factor-β Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Garidou, L.; Heydari, S.; Gossa, S.; McGavern, D.B. Therapeutic Blockade of Transforming Growth Factor Beta Fails To Promote Clearance of a Persistent Viral Infection. J. Virol. 2012, 86, 7060–7071. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-Intrinsic Transforming Growth Factor-β Signaling Mediates Virus-Specific CD8+ T Cell Deletion and Viral Persistence In Vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.M.; Wehrens, E.J.; Labarta-Bajo, L.; Streeck, H.; Zuniga, E.I. TGF-β receptor maintains CD4 T helper cell identity during chronic viral infections. J. Clin. Investig. 2016, 126, 3799–3813. [Google Scholar] [CrossRef] [PubMed]
- Boettler, T.; Cheng, Y.; Ehrhardt, K.; von Herrath, M. TGF-β Blockade Does Not Improve Control of an Established Persistent Viral Infection. Viral Immunol. 2012, 25, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Wolint, P.; Walton, S.; Schwarz, K.; Oxenius, A. Differential role of IL-2R signaling for CD8+ T cell responses in acute and chronic viral infections. Eur. J. Immunol. 2007, 37, 1502–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattman, J.N.; Grayson, J.M.; Wherry, E.J.; Kaech, S.M.; Smith, K.A.; Ahmed, R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 2003, 9, 540–547. [Google Scholar] [CrossRef]
- Beltra, J.-C.; Bourbonnais, S.; Bédard, N.; Charpentier, T.; Boulangé, M.; Michaud, E.; Boufaied, I.; Bruneau, J.; Shoukry, N.H.; Lamarre, A.; et al. IL2Rβ-dependent signals drive terminal exhaustion and suppress memory development during chronic viral infection. Proc. Natl. Acad. Sci. USA 2016, 113, E5444–E5453. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.J.; Hildeman, D.A.; Sabbaj, S.; Gaddis, D.E.; Tebo, A.E.; Shang, L.; Goepfert, P.A.; Zajac, A.J. Cutting edge: emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J. Immunol. 2005, 174, 5926–5930. [Google Scholar] [CrossRef]
- Lang, K.S.; Recher, M.; Navarini, A.A.; Harris, N.L.; Löhning, M.; Junt, T.; Probst, H.C.; Hengartner, H.; Zinkernagel, R.M. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur. J. Immunol. 2005, 35, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Elsaesser, H.; Sauer, K.; Brooks, D.G. IL-21 Is Required to Control Chronic Viral Infection. Science 2009, 324, 1569–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, A.; Kisielow, J.; Schmitz, I.; Freigang, S.; Shamshiev, A.T.; Weber, J.; Marsland, B.J.; Oxenius, A.; Kopf, M. IL-21R on T Cells Is Critical for Sustained Functionality and Control of Chronic Viral Infection. Science 2009, 324, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Du, M.; Zajac, A.J. A vital role for interleukin-21 in the control of a chronic viral infection. Science 2009, 324, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Jandl, C.; Liu, S.M.; Cañete, P.F.; Warren, J.; Hughes, W.E.; Vogelzang, A.; Webster, K.; Craig, M.E.; Uzel, G.; Dent, A.; et al. IL-21 restricts T follicular regulatory T cell proliferation through Bcl-6 mediated inhibition of responsiveness to IL-2. Nat. Commun. 2017, 8, 14647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, M.A.U.; Latner, D.R.; Aubert, R.D.; Gourley, T.; Spolski, R.; Davis, C.W.; Langley, W.A.; Ha, S.-J.; Ye, L.; Sarkar, S.; et al. Interleukin-21 Is a Critical Cytokine for the Generation of Virus-Specific Long-Lived Plasma Cells. J. Virol. 2013, 87, 7737–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kahan, S.M.; Zajac, A.J. Immune Exhaustion: Past Lessons and New Insights from Lymphocytic Choriomeningitis Virus. Viruses 2019, 11, 156. https://doi.org/10.3390/v11020156
Kahan SM, Zajac AJ. Immune Exhaustion: Past Lessons and New Insights from Lymphocytic Choriomeningitis Virus. Viruses. 2019; 11(2):156. https://doi.org/10.3390/v11020156
Chicago/Turabian StyleKahan, Shannon M., and Allan J. Zajac. 2019. "Immune Exhaustion: Past Lessons and New Insights from Lymphocytic Choriomeningitis Virus" Viruses 11, no. 2: 156. https://doi.org/10.3390/v11020156
APA StyleKahan, S. M., & Zajac, A. J. (2019). Immune Exhaustion: Past Lessons and New Insights from Lymphocytic Choriomeningitis Virus. Viruses, 11(2), 156. https://doi.org/10.3390/v11020156