Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
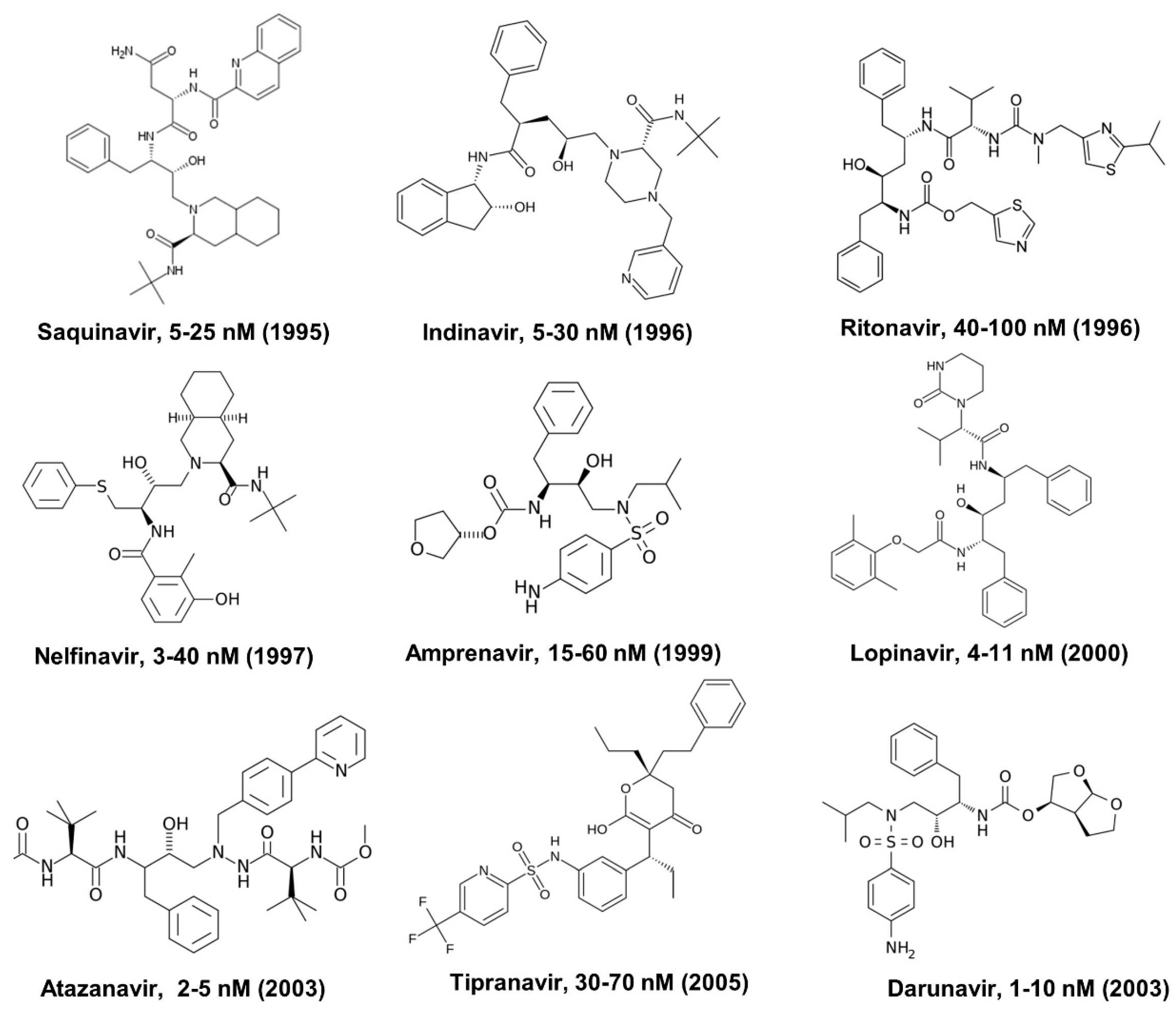
2. Human Immunodeficiency Virus (HIV) Protease Inhibitors
3. Hepatitis C Virus (HCV) Protease Inhibitors
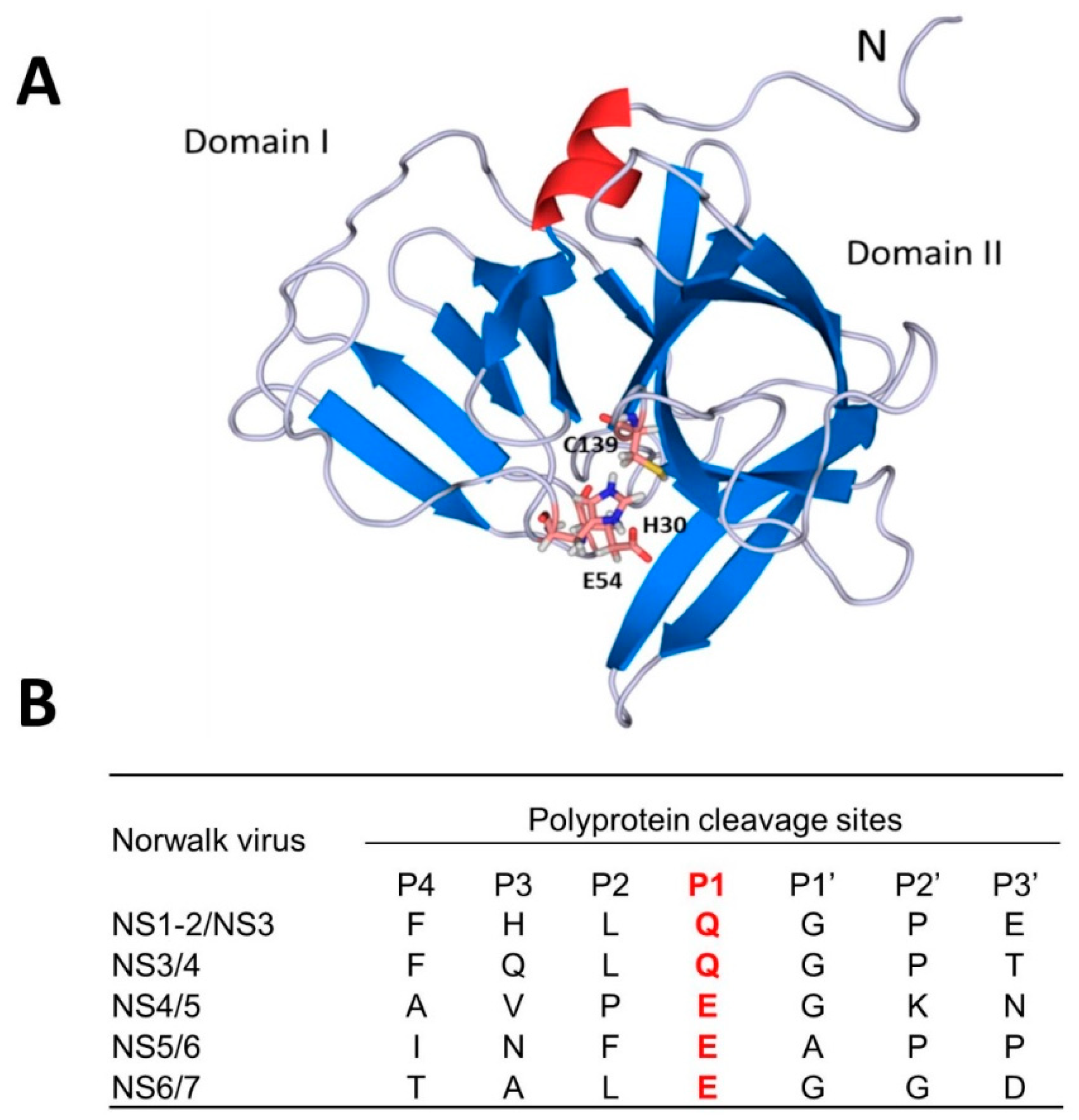
4. Norovirus 3CL Protease (NV 3CLPro) Inhibitors
4.1. Structure-Guided Optimization of Dipeptidyl Inhibitor Lead Series
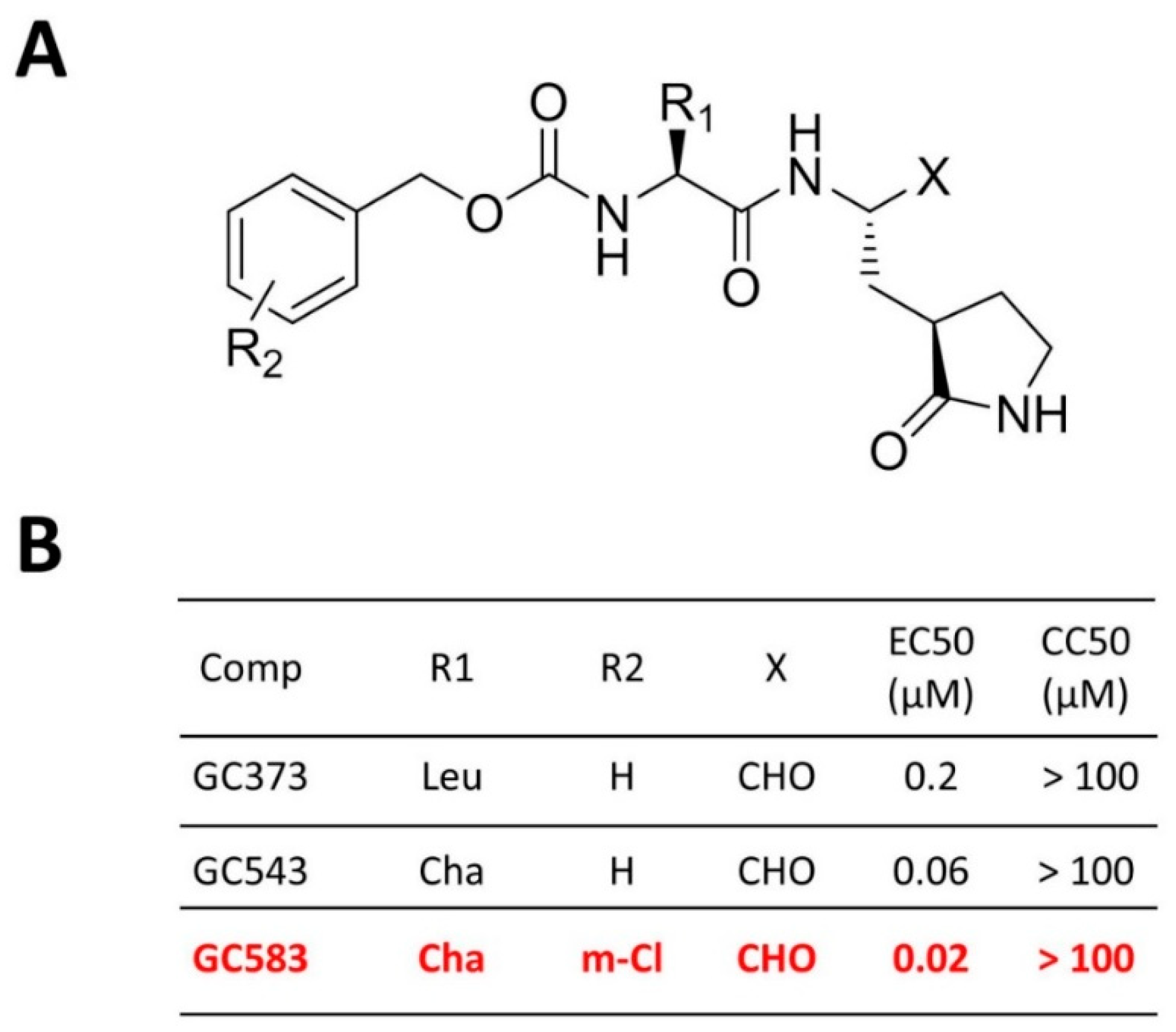
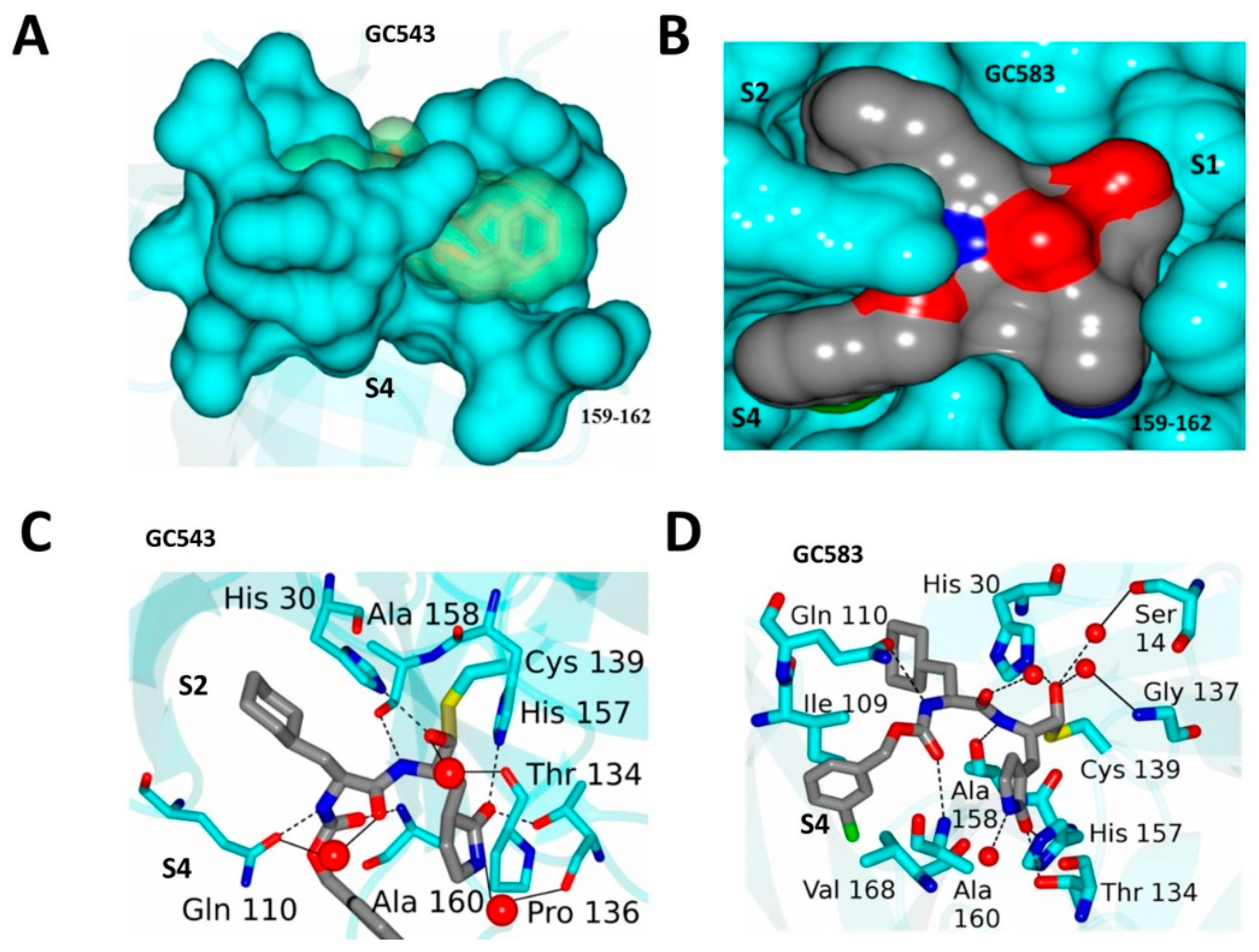
4.2. Optimization at the P2 and P3 Positions
4.3. Prodrug Approach
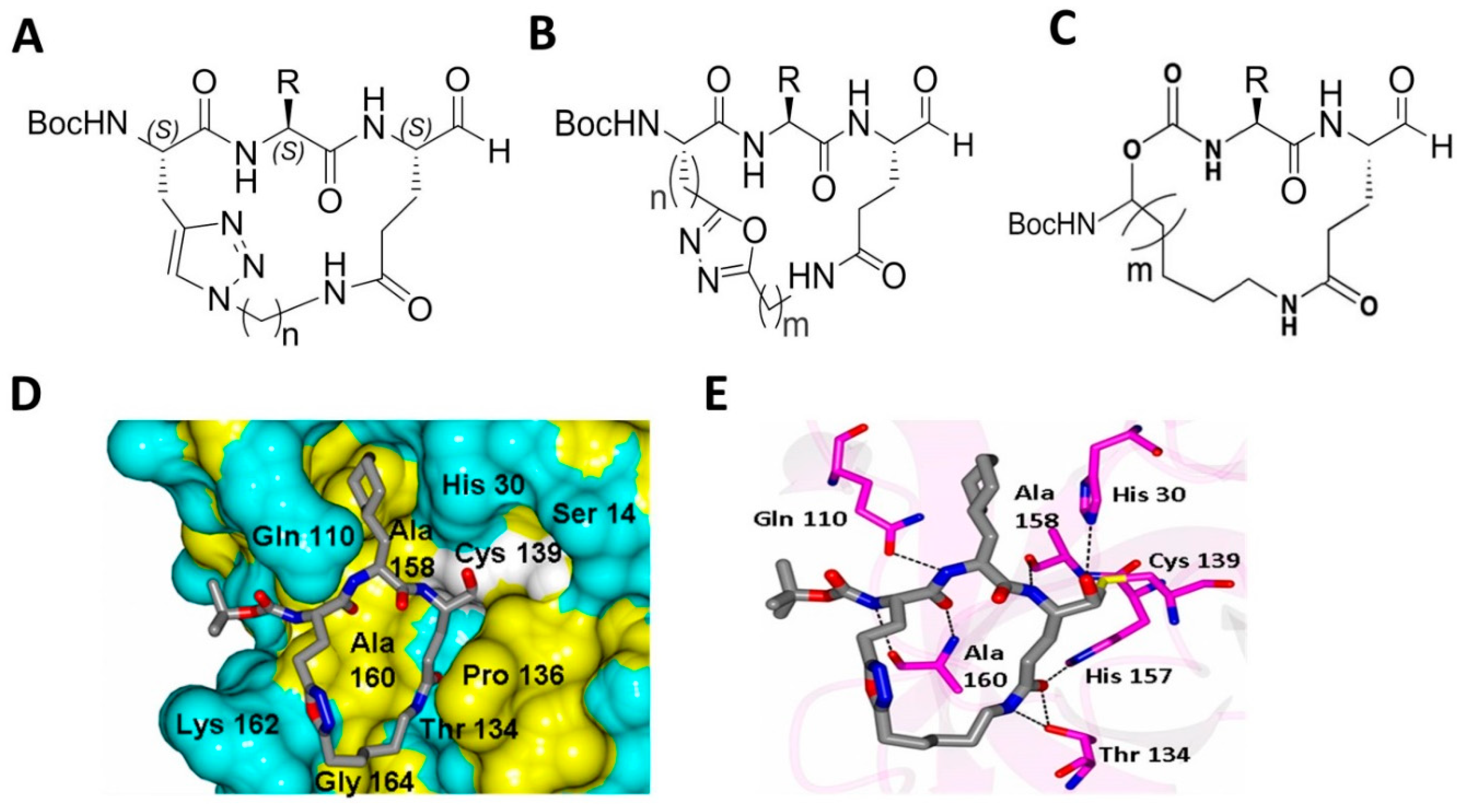
4.4. Macrocyclic Inhibitors Targeting Norovirus 3CLPro
4.5. Tripeptidyl Inhibitors
4.6. Potential of Dipeptidyl Compounds as Antiviral Drugs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Puente: X., S.; Sanchez, L.M.; Overall, C.M.; Lopez-Otin, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
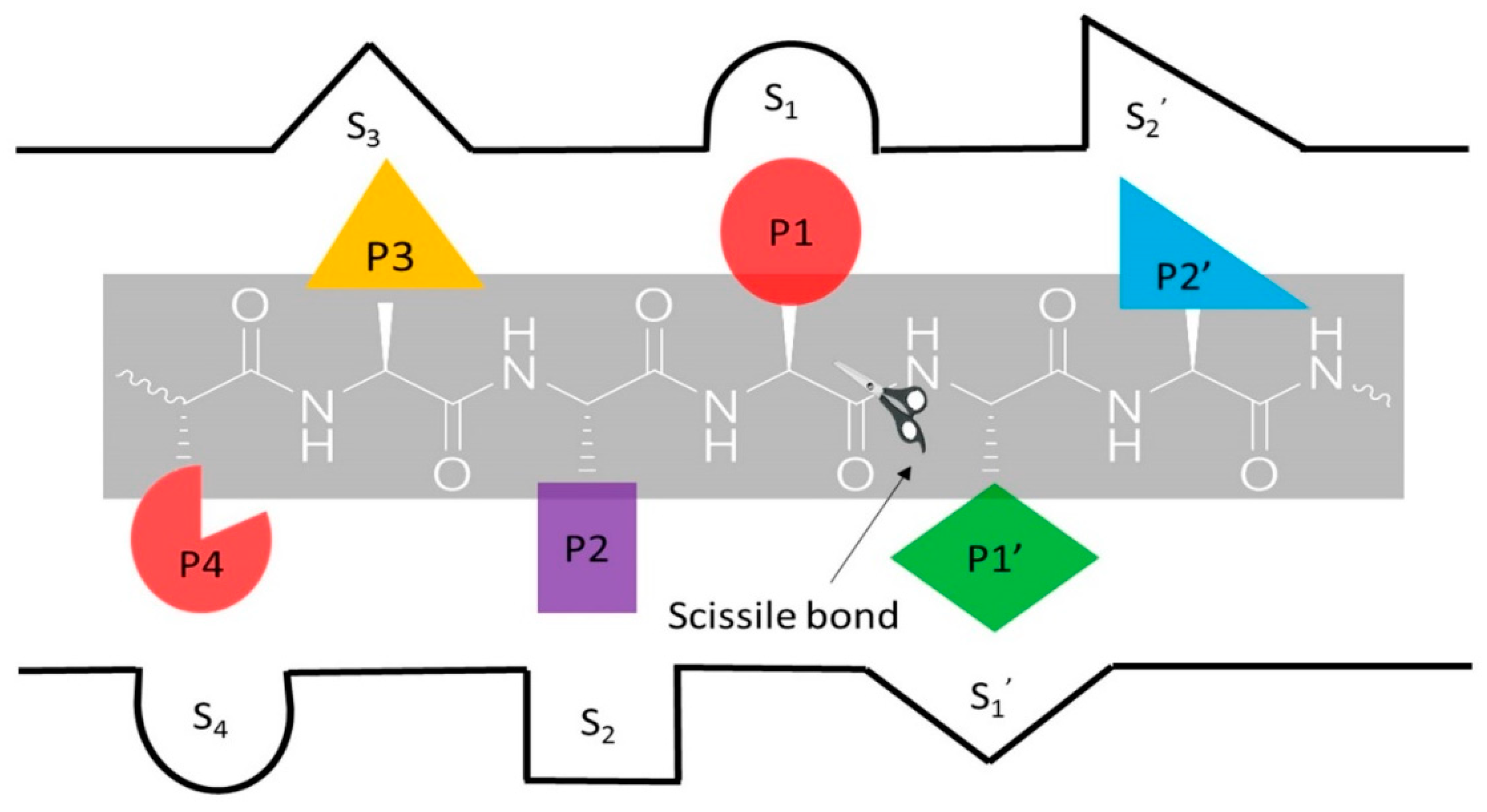
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Huber, R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 1992, 204, 433–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otlewski, J.; Krowarsch, D.; Apostoluk, W. Protein inhibitors of serine proteinases. Acta Biochim. Pol. 1999, 46, 531–565. [Google Scholar] [PubMed]
- Miravitlles, M. Alpha-1-antitrypsin and other proteinase inhibitors. Curr. Opin. Pharmacol. 2012, 12, 309–314. [Google Scholar] [CrossRef] [PubMed]
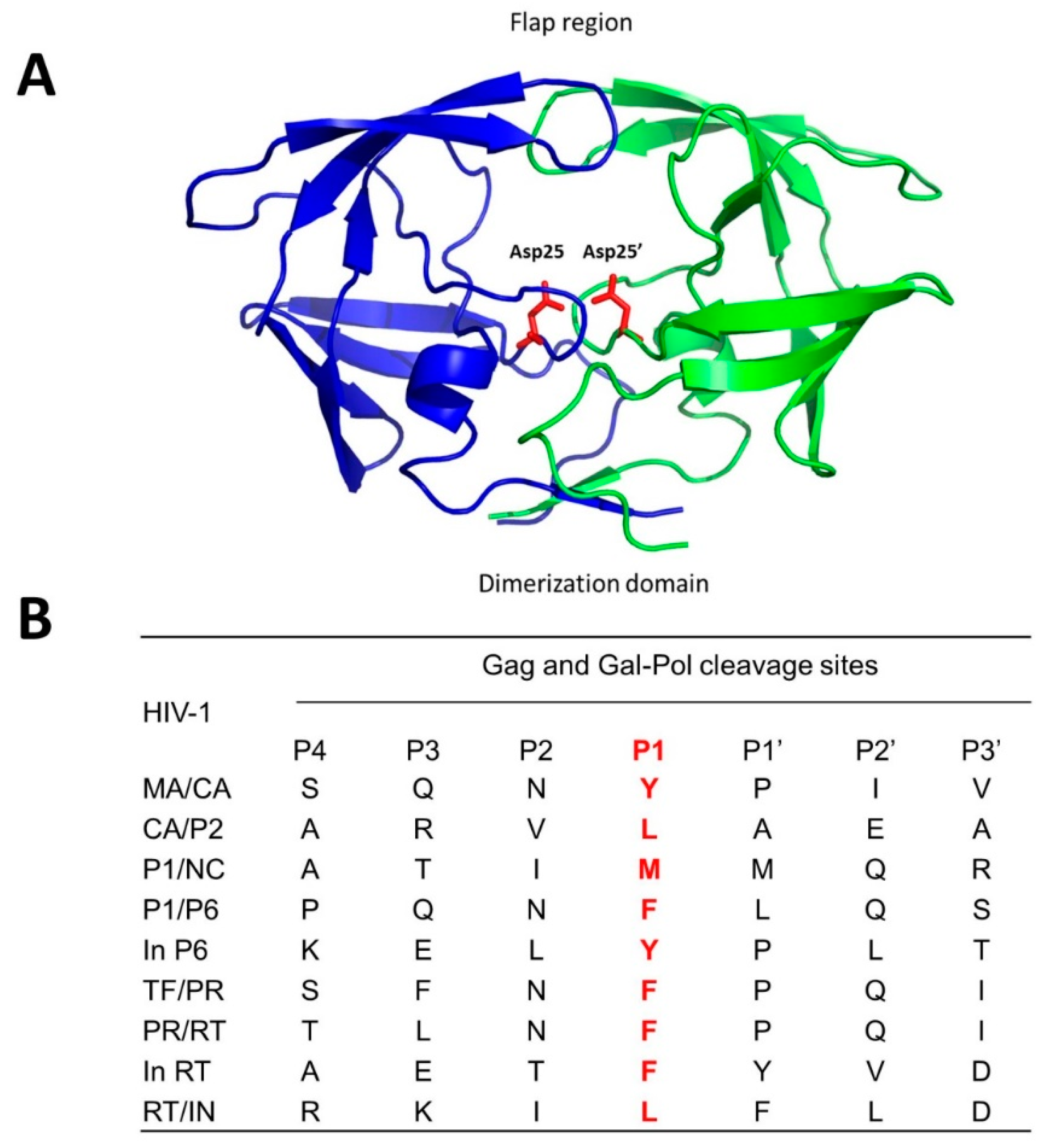
- Brik, A.; Wong, C.H. HIV-1 protease: Mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Beck, Z.Q.; Lin, Y.C.; Elder, J.H. Molecular basis for the relative substrate specificity of human immunodeficiency virus type 1 and feline immunodeficiency virus proteases. J. Virol. 2001, 75, 9458–9469. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Henderson, G.J.; Schiffer, C.A.; Swanstrom, R. Replacement of the P1 amino acid of human immunodeficiency virus type 1 Gag processing sites can inhibit or enhance the rate of cleavage by the viral protease. J. Virol. 2002, 76, 10226–10233. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl.) 2015, 7, 95–104. [Google Scholar] [PubMed]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.; Nalivaika, E.A.; Ozen, A.; et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbaiah, M.A.M.; Meanwell, N.A.; Kadow, J.F. Design strategies in the prodrugs of HIV-1 protease inhibitors to improve the pharmaceutical properties. Eur. J. Med. Chem. 2017, 139, 865–883. [Google Scholar] [CrossRef] [PubMed]
- Germer, J.J.; Mandrekar, J.N.; Bendel, J.L.; Mitchell, P.S.; Yao, J.D. Hepatitis C virus genotypes in clinical specimens tested at a national reference testing laboratory in the United States. J. Clin. Microbiol. 2011, 49, 3040–3043. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Morgenstern, K.A.; Lin, C.; Fox, T.; Dwyer, M.D.; Landro, J.A.; Chambers, S.P.; Markland, W.; Lepre, C.A.; O’Malley, E.T.; et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996, 87, 343–355. [Google Scholar] [CrossRef]
- Love, R.A.; Parge, H.E.; Wickersham, J.A.; Hostomsky, Z.; Habuka, N.; Moomaw, E.W.; Adachi, T.; Hostomska, Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 1996, 87, 331–342. [Google Scholar] [CrossRef]
- Urbani, A.; Bianchi, E.; Narjes, F.; Tramontano, A.; de Francesco, R.; Steinkuhler, C.; Pessi, A. Substrate specificity of the hepatitis C virus serine protease NS3. J. Biol. Chem. 1997, 272, 9204–9209. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Kauffman, R.S.; Hurter, P.; Mueller, P. Discovery and development of telaprevir: An NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 2011, 29, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.; Venkatraman, S. The Discovery and Development of Boceprevir: A Novel, First-generation Inhibitor of the Hepatitis C Virus NS3/4A Serine Protease. J. Clin. Transl. Hepatol. 2013, 1, 22–32. [Google Scholar] [PubMed] [Green Version]
- McCauley, J.A.; Rudd, M.T. Hepatitis C virus NS3/4a protease inhibitors. Curr. Opin. Pharmacol. 2016, 30, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Perni, R.B.; Pitlik, J.; Britt, S.D.; Court, J.J.; Courtney, L.F.; Deininger, D.D.; Farmer, L.J.; Gates, C.A.; Harbeson, S.L.; Levin, R.B.; et al. Inhibitors of hepatitis C virus NS3.4A protease 2. Warhead SAR and optimization. Bioorg. Med. Chem. Lett. 2004, 14, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef] [PubMed]
- Green, K.Y. Caliciviridae: The Noroviruses, 6th ed.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Pringle, K.; Lopman, B.; Vega, E.; Vinje, J.; Parashar, U.D.; Hall, A.J. Noroviruses: Epidemiology, immunity and prospects for prevention. Future Microbiol. 2015, 10, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Phan, K.; Teng, I.; Pu, J.; Watanabe, T. A systematic review and meta-analysis of the prevalence of norovirus in cases of gastroenteritis in developing countries. Medicine (Baltimore) 2017, 96, e8139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galasiti Kankanamalage, A.C.; Kim, Y.; Rathnayake, A.D.; Alliston, K.R.; Butler, M.M.; Cardinale, S.C.; Bowlin, T.L.; Groutas, W.C.; Chang, K.O. Design, Synthesis, and Evaluation of Novel Prodrugs of Transition State Inhibitors of Norovirus 3CL Protease. J. Med. Chem. 2017, 60, 6239–6248. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.D.; Goulter, R.M.; Jaykus, L.A. Human norovirus as a foodborne pathogen: Challenges and developments. Annu. Rev. Food Sci. Technol. 2015, 6, 411–433. [Google Scholar] [CrossRef] [PubMed]
- Bok, K.; Green, K.Y. Norovirus gastroenteritis in immunocompromised patients. N. Engl. J. Med. 2012, 367, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.P.; Hall, A.J. Norovirus Illnesses in Children and Adolescents. Infect. Dis. Clin. North Am. 2018, 32, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; King, A.D.; Green, K.Y. Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology 2006, 353, 463–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wobus, C.E.; Karst, S.M.; Thackray, L.B.; Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Krug, A.; Mackenzie, J.M.; Green, K.Y.; Virgin, H.W. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004, 2, e432. [Google Scholar] [CrossRef] [PubMed]
- Tiew, K.C.; He, G.; Aravapalli, S.; Mandadapu, S.R.; Gunnam, M.R.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Chang, K.O.; Groutas, W.C. Design, synthesis, and evaluation of inhibitors of Norwalk virus 3C protease. Bioorg. Med. Chem. Lett. 2011, 21, 5315–5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandadapu, S.R.; Weerawarna, P.M.; Gunnam, M.R.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Chang, K.O.; Groutas, W.C. Potent inhibition of norovirus 3CL protease by peptidyl alpha-ketoamides and alpha-ketoheterocycles. Bioorg. Med. Chem. Lett. 2012, 22, 4820–4826. [Google Scholar] [CrossRef] [PubMed]
- Mandadapu, S.R.; Gunnam, M.R.; Galasiti Kankanamalage, A.C.; Uy, R.A.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Chang, K.O.; Groutas, W.C. Potent inhibition of norovirus by dipeptidyl alpha-hydroxyphosphonate transition state mimics. Bioorg. Med. Chem. Lett. 2013, 23, 5941–5944. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.O.; Takahashi, D.; Prakash, O.; Kim, Y. Characterization and inhibition of norovirus proteases of genogroups I and II using a fluorescence resonance energy transfer assay. Virology 2012, 423, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandadapu, S.R.; Gunnam, M.R.; Tiew, K.C.; Uy, R.A.; Prior, A.M.; Alliston, K.R.; Hua, D.H.; Kim, Y.; Chang, K.O.; Groutas, W.C. Inhibition of norovirus 3CL protease by bisulfite adducts of transition state inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 62–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galasiti Kankanamalage, A.C.; Kim, Y.; Weerawarna, P.M.; Uy, R.A.; Damalanka, V.C.; Mandadapu, S.R.; Alliston, K.R.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; et al. Structure-guided design and optimization of dipeptidyl inhibitors of norovirus 3CL protease. Structure-activity relationships and biochemical, X-ray crystallographic, cell-based, and in vivo studies. J. Med. Chem. 2015, 58, 3144–3155. [Google Scholar] [CrossRef] [PubMed]
- Galasiti Kankanamalage, A.C.; Kim, Y.; Rathnayake, A.D.; Damalanka, V.C.; Weerawarna, P.M.; Doyle, S.T.; Alsoudi, A.F.; Dissanayake, D.M.P.; Lushington, G.H.; Mehzabeen, N.; et al. Structure-based exploration and exploitation of the S4 subsite of norovirus 3CL protease in the design of potent and permeable inhibitors. Eur. J. Med. Chem. 2017, 126, 502–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madala, P.K.; Tyndall, J.D.A.; Nall, T.; Fairlie, D.P. Update 1 of: proteases universally recognize beta strands in their active sites. Chem. Rev. 2010, 110, 3299–3314. [Google Scholar] [CrossRef] [PubMed]
- Glenn, M.P.; Pattenden, L.K.; Reid, R.C.; Tyssen, D.P.; Tyndall, J.D.; Birch, C.J.; Fairlie, D.P. Beta-strand mimicking macrocyclic amino acids: Templates for protease inhibitors with antiviral activity. J. Med. Chem. 2002, 45, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Marsault, E.; Peterson, M.L. Macrocycles are great cycles: Applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 2011, 54, 1961–2004. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Mandadapu, S.R.; Weerawarna, P.M.; Prior, A.M.; Uy, R.A.; Aravapalli, S.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Hua, D.H.; Chang, K.O.; et al. Macrocyclic inhibitors of 3C and 3C-like proteases of picornavirus, norovirus, and coronavirus. Bioorg. Med. Chem. Lett. 2013, 23, 3709–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerawarna, P.M.; Kim, Y.; Galasiti Kankanamalage, A.C.; Damalanka, V.C.; Lushington, G.H.; Alliston, K.R.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Chang, K.O.; et al. Structure-based design and synthesis of triazole-based macrocyclic inhibitors of norovirus protease: Structural, biochemical, spectroscopic, and antiviral studies. Eur. J. Med. Chem. 2016, 119, 300–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damalanka, V.C.; Kim, Y.; Galasiti Kankanamalage, A.C.; Lushington, G.H.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Chang, K.O.; Groutas, W.C. Design, synthesis, and evaluation of a novel series of macrocyclic inhibitors of norovirus 3CL protease. Eur. J. Med. Chem. 2017, 127, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Damalanka, V.C.; Kim, Y.; Alliston, K.R.; Weerawarna, P.M.; Galasiti Kankanamalage, A.C.; Lushington, G.H.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Chang, K.O.; et al. Oxadiazole-Based Cell Permeable Macrocyclic Transition State Inhibitors of Norovirus 3CL Protease. J. Med. Chem. 2016, 59, 1899–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binford, S.L.; Maldonado, F.; Brothers, M.A.; Weady, P.T.; Zalman, L.S.; Meador, J.W.; Matthews, D.A.; Patick, A.K. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 2005, 49, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Prior, A.M.; Kim, Y.; Weerasekara, S.; Moroze, M.; Alliston, K.R.; Uy, R.A.; Groutas, W.C.; Chang, K.O.; Hua, D.H. Design, synthesis, and bioevaluation of viral 3C and 3C-like protease inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6317–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Muhaxhiri, Z.; Estes, M.K.; Palzkill, T.; Prasad, B.V.; Song, Y. Synthesis, Activity and Structure-Activity Relationship of Noroviral Protease Inhibitors. MedChemComm 2013, 4, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Amblard, F.; Zhou, S.; Liu, P.; Yoon, J.; Cox, B.; Muzzarelli, K.; Kuiper, B.D.; Kovari, L.C.; Schinazi, R.F. Synthesis and antiviral evaluation of novel peptidomimetics as norovirus protease inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Mandadapu, S.R.; Groutas, W.C.; Chang, K.O. Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antiviral. Res. 2013, 97, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Weerasekara, S.; Hua, D.H.; Groutas, W.C.; Chang, K.O.; Pedersen, N.C. Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 2016, 12, e1005531. [Google Scholar]
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.O. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, K.-O.; Kim, Y.; Lovell, S.; Rathnayake, A.D.; Groutas, W.C. Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors. Viruses 2019, 11, 197. https://doi.org/10.3390/v11020197
Chang K-O, Kim Y, Lovell S, Rathnayake AD, Groutas WC. Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors. Viruses. 2019; 11(2):197. https://doi.org/10.3390/v11020197
Chicago/Turabian StyleChang, Kyeong-Ok, Yunjeong Kim, Scott Lovell, Athri D. Rathnayake, and William C. Groutas. 2019. "Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors" Viruses 11, no. 2: 197. https://doi.org/10.3390/v11020197
APA StyleChang, K. -O., Kim, Y., Lovell, S., Rathnayake, A. D., & Groutas, W. C. (2019). Antiviral Drug Discovery: Norovirus Proteases and Development of Inhibitors. Viruses, 11(2), 197. https://doi.org/10.3390/v11020197