Spread of Highly Pathogenic Avian Influenza (HPAI) H5N5 Viruses in Europe in 2016–2017 Appears Related to the Timing of Reassortment Events


Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Detection and Sequencing
2.2. Phylogenetic Analysis
2.3. Network Analysis
2.4. Molecular Clock Analysis
2.5. Cell Cultures
2.6. Virus Propagation and Titration
2.7. Virus Infection of Primary Chicken and Duck Cells
3. Results
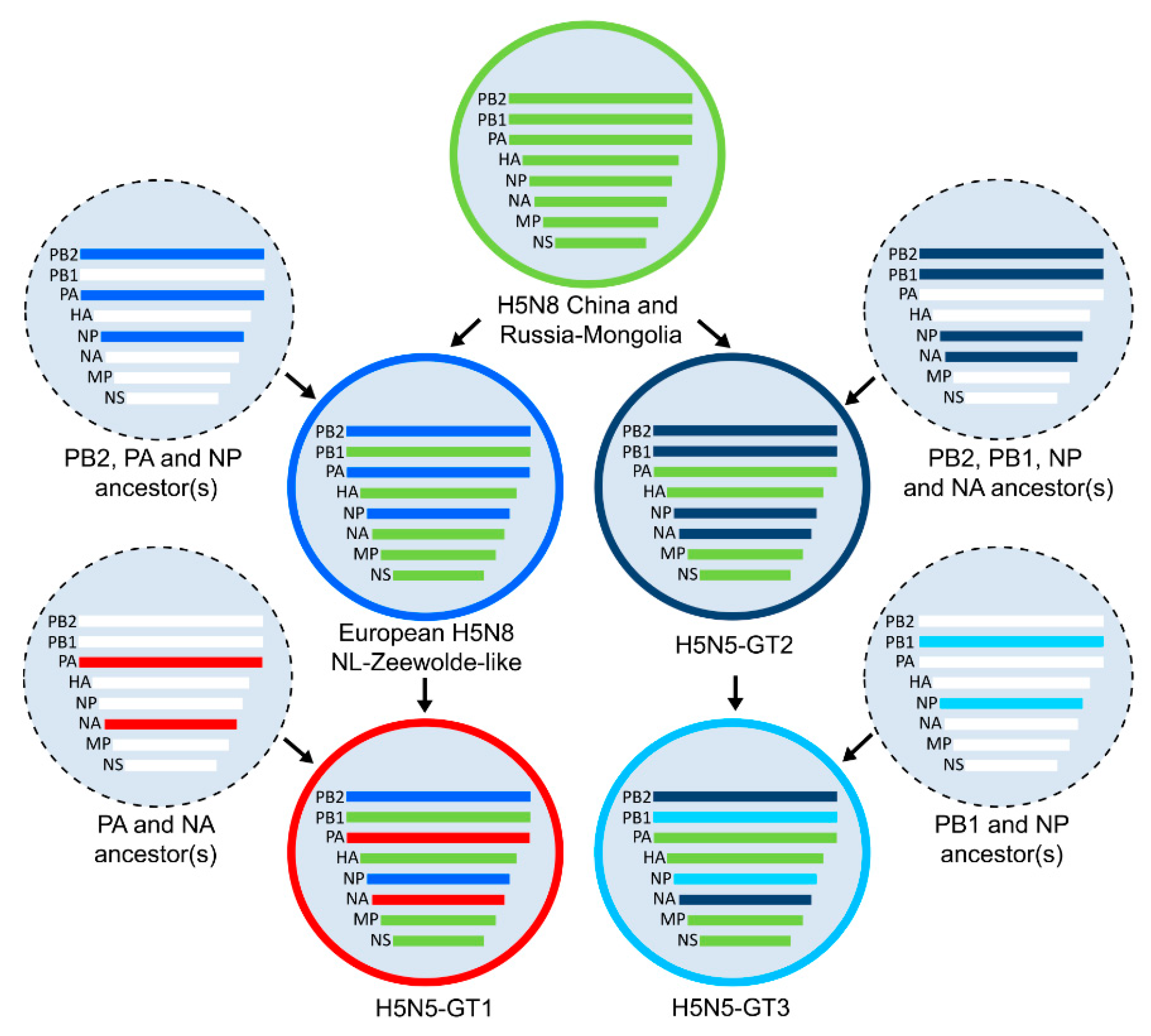
3.1. Genetic Analysis of HPAI H5N5 Viruses in the Netherlands
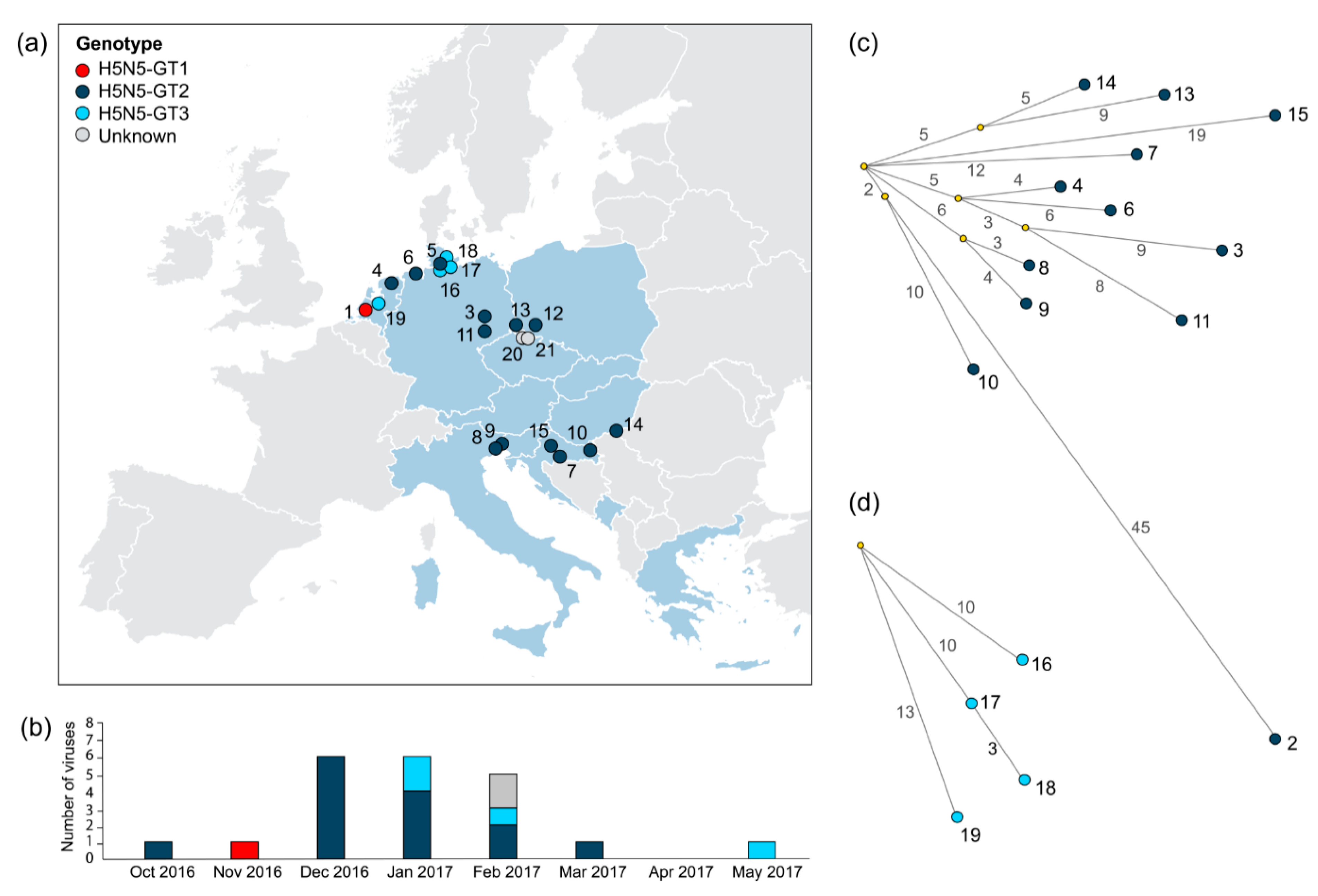
3.2. Incidence and Spatiotemporal Distribution of HPAI H5N5 Genotypes in Europe
3.3. Genetic Relationships between HPAI H5N5 Viruses
3.4. Timing of the HPAI H5N5 Reassortment Events
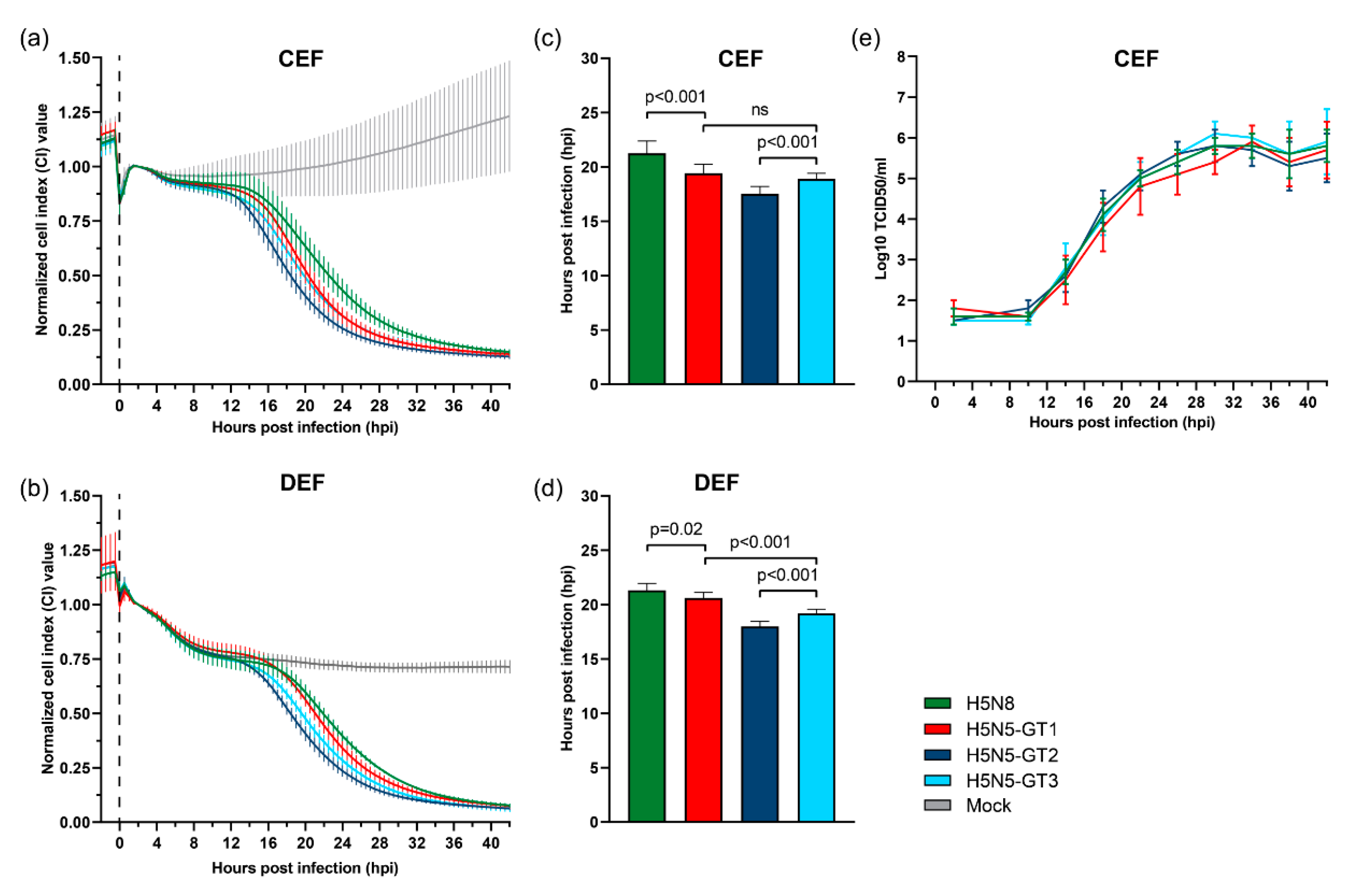
3.5. Cytopathogenicity and Replication of HPAI H5N5 Viruses in Primary Chicken and Duck Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza a/goose/Guangdong/1/96 (H5N1) virus: Similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.K. Outbreak of avian influenza A (H5N1) virus infection in Hong Kong in 1997. Clin. Infect. Dis. 2002, 34 (Suppl. 2), S58–S64. [Google Scholar] [CrossRef]
- Ellis, T.M.; Bousfield, R.B.; Bissett, L.A.; Dyrting, K.C.; Luk, G.S.; Tsim, S.T.; Sturm-Ramirez, K.; Webster, R.G.; Guan, Y.; Malik Peiris, J.S. Investigation of outbreaks of highly pathogenic H5N1 avian influenza in waterfowl and wild birds in Hong Kong in late 2002. Avian Pathol. 2004, 33, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza A virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Peng, X.; Xu, L.; Jin, C.; Cheng, L.; Lu, X.; Xie, T.; Yao, H.; Wu, N. Novel reassortant influenza A (H5N8) viruses in domestic ducks, Eastern China. Emerg. Infect. Dis. 2014, 20, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kang, H.M.; Lee, E.K.; Song, B.M.; Jeong, J.; Kwon, Y.K.; Kim, H.R.; Lee, K.J.; Hong, M.S.; Jang, I.; et al. Novel reassortant influenza a (H5N8) viruses, South Korea, 2014. Emerg. Infect. Dis. 2014, 20, 1087–1089. [Google Scholar] [CrossRef] [PubMed]
- Bouwstra, R.; Koch, G.; Heutink, R.; Harders, F.; van der Spek, A.; Elbers, A.R.; Bossers, A. Phylogenetic analysis of highly pathogenic avian influenza A (H5N8) virus outbreak strains provides evidence for four separate introductions and one between-poultry farm transmission in the Netherlands, November 2014. Euro. Surveill. 2015, 20, 21174. [Google Scholar] [CrossRef]
- Harder, T.; Maurer-Stroh, S.; Pohlmann, A.; Starick, E.; Horeth-Bontgen, D.; Albrecht, K.; Pannwitz, G.; Teifke, J.; Gunalan, V.; Lee, R.T.; et al. Influenza A (H5N8) virus similar to strain in Korea causing highly pathogenic avian influenza in Germany. Emerg. Infect. Dis. 2015, 21, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Torchetti, M.K.; Winker, K.; Ip, H.S.; Song, C.S.; Swayne, D.E. Intercontinental spread of Asian-origin H5N8 to north America through Beringia by migratory birds. J. Virol. 2015, 89, 6521–6524. [Google Scholar] [CrossRef] [PubMed]
- Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. In Science; AAAS: Washington, DC, USA, 2016; Volume 354, pp. 213–217. [Google Scholar]
- Lee, D.H.; Bahl, J.; Torchetti, M.K.; Killian, M.L.; Ip, H.S.; DeLiberto, T.J.; Swayne, D.E. Highly pathogenic avian influenza viruses and generation of novel reassortants, United States, 2014–2015. Emerg. Infect. Dis. 2016, 22, 1283–1285. [Google Scholar] [CrossRef]
- Li, M.; Liu, H.; Bi, Y.; Sun, J.; Wong, G.; Liu, D.; Li, L.; Liu, J.; Chen, Q.; Wang, H.; et al. Highly pathogenic avian influenza A (H5N8) virus in wild migratory birds, Qinghai Lake, China. Emerg. Infect. Dis. 2017, 23, 637–641. [Google Scholar] [CrossRef]
- Lee, D.H.; Sharshov, K.; Swayne, D.E.; Kurskaya, O.; Sobolev, I.; Kabilov, M.; Alekseev, A.; Irza, V.; Shestopalov, A. Novel reassortant clade 2.3.4.4 avian influenza a (H5N8) virus in wild aquatic birds, Russia, 2016. Emerg. Infect. Dis. 2017, 23, 359–360. [Google Scholar] [CrossRef]
- Marchenko, V.Y.; Susloparov, I.M.; Komissarov, A.B.; Fadeev, A.; Goncharova, N.I.; Shipovalov, A.V.; Svyatchenko, S.V.; Durymanov, A.G.; Ilyicheva, T.N.; Salchak, L.K.; et al. Reintroduction of highly pathogenic avian influenza A/ H5N8 virus of clade 2.3.4.4. In Russia. Arch. Virol. 2017, 162, 1381–1385. [Google Scholar] [CrossRef]
- Lee, D.H.; Bertran, K.; Kwon, J.H.; Swayne, D.E. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Napp, S.; Majo, N.; Sanchez-Gonzalez, R.; Vergara-Alert, J. Emergence and spread of highly pathogenic avian influenza a (H5N8) in Europe in 2016–2017. Transbound. Emerg. Dis. 2018, 65, 1217–1226. [Google Scholar] [CrossRef]
- Kwon, J.H.; Jeong, S.; Lee, D.H.; Swayne, D.E.; Kim, Y.J.; Lee, S.H.; Noh, J.Y.; Erdene-Ochir, T.O.; Jeong, J.H.; Song, C.S. New reassortant clade 2.3.4.4b avian influenza a (H5N6) virus in wild birds, South Korea, 2017–2018. Emerg. Infect. Dis. 2018, 24, 1953–1955. [Google Scholar] [CrossRef] [PubMed]
- Beerens, N.; Koch, G.; Heutink, R.; Harders, F.; Vries, D.P.E.; Ho, C.; Bossers, A.; Elbers, A. Novel highly pathogenic avian influenza a (H5N6) virus in the Netherlands, December 2017. Emerg. Infect. Dis. 2018, 24, 770. [Google Scholar] [CrossRef]
- Brown, I.; Mulatti, P.; Smietanka, K.; Staubach, C.; Willeberg, P.; Adlhoch, C.; Candiani, D.; Fabris, C.; Zancanaro, G.; Morgado, J.; et al. Avian influenza overview October 2016–August 2017. EFSA J. 2017, 15, 5018. [Google Scholar]
- Marchenko, V.; Goncharova, N.; Susloparov, I.; Kolosova, N.; Gudymo, A.; Svyatchenko, S.; Danilenko, A.; Durymanov, A.; Gavrilova, E.; Maksyutov, R.; et al. Isolation and characterization of H5Nx highly pathogenic avian influenza viruses of clade 2.3.4.4 in Russia. Virology 2018, 525, 216–223. [Google Scholar] [CrossRef]
- Beerens, N.; Heutink, R.; Bergervoet, S.A.; Harders, F.; Bossers, A.; Koch, G. Multiple reassorted viruses as cause of highly pathogenic avian influenza a (H5N8) virus epidemic, the Netherlands, 2016. Emerg. Infect. Dis. 2017, 23, 1974–1981. [Google Scholar] [CrossRef]
- Fusaro, A.; Monne, I.; Mulatti, P.; Zecchin, B.; Bonfanti, L.; Ormelli, S.; Milani, A.; Cecchettin, K.; Lemey, P.; Moreno, A.; et al. Genetic diversity of highly pathogenic avian influenza a (H5N8/H5N5) viruses in Italy, 2016–2017. Emerg. Infect. Dis. 2017, 23, 1543–1547. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, A.; Starick, E.; Grund, C.; Hoper, D.; Strebelow, G.; Globig, A.; Staubach, C.; Conraths, F.J.; Mettenleiter, T.C.; Harder, T.; et al. Swarm incursions of reassortants of highly pathogenic avian influenza virus strains H5N8 and H5N5, clade 2.3.4.4b, Germany, Winter 2016/17. Sci. Rep. 2018, 8, 15. [Google Scholar] [CrossRef]
- Savic, V. Novel reassortant clade 2.3.4.4 avian influenza a (H5N5) virus in wild birds and poultry, Croatia, 2016–2017. Veterinarski. Arhiv. 2017, 87, 377–396. [Google Scholar] [CrossRef]
- Nagy, A.; Dan, A.; Cernikova, L.; Vitaskova, E.; Krivda, V.; Hornickova, J.; Masopust, R.; Sedlak, K. Microevolution and independent incursions as main forces shaping h5 hemagglutinin diversity during a H5N8/H5N5 highly pathogenic avian influenza outbreak in Czech Republic in 2017. Arch. Virol. 2018, 163, 2219–2224. [Google Scholar] [CrossRef]
- Swieton, E.; Smietanka, K. Phylogenetic and molecular analysis of highly pathogenic avian influenza H5N8 and H5N5 viruses detected in Poland in 2016–2017. Transbound. Emerg. Dis. 2018, 65, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, R.A.; Bestebroer, T.M.; Herfst, S.; Van Der Kemp, L.; Rimmelzwaan, G.F.; Osterhaus, A.D. Detection of influenza a viruses from different species by PCR amplification of conserved sequences in the matrix gene. J. Clin. Microbiol. 2000, 38, 4096–4101. [Google Scholar] [PubMed]
- Slomka, M.J.; Pavlidis, T.; Banks, J.; Shell, W.; McNally, A.; Essen, S.; Brown, I.H. Validated H5 Eurasian real-time reverse transcriptase-polymerase chain reaction and its application in H5N1 outbreaks in 2005–2006. Avian Dis. 2007, 51, 373–377. [Google Scholar] [CrossRef]
- Slomka, M.J.; Pavlidis, T.; Coward, V.J.; Voermans, J.; Koch, G.; Hanna, A.; Banks, J.; Brown, I.H. Validated realtime reverse transcriptase PCR methods for the diagnosis and pathotyping of Eurasian H7 avian influenza viruses. Influenza Other Respir. Viruses 2009, 3, 151–164. [Google Scholar] [CrossRef]
- Gall, A.; Hoffmann, B.; Harder, T.; Grund, C.; Beer, M. Universal primer set for amplification and sequencing of HA0 cleavage sites of all influenza a viruses. J. Clin. Microbiol. 2008, 46, 2561–2567. [Google Scholar] [CrossRef]
- Gall, A.; Hoffmann, B.; Harder, T.; Grund, C.; Ehricht, R.; Beer, M. Rapid and highly sensitive neuraminidase subtyping of avian influenza viruses by use of a diagnostic DNA microarray. J. Clin. Microbiol. 2009, 47, 2985–2988. [Google Scholar] [CrossRef]
- Watson, S.J.; Welkers, M.R.; Depledge, D.P.; Coulter, E.; Breuer, J.M.; de Jong, M.D.; Kellam, P. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013, 368, 20120205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—from vision to reality. Euro. Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. Cd-hit: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Larsson, A. Aliview: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef]
- Van Zaane, D.; Brinkhof, J.M.; Westenbrink, F.; Gielkens, A.L. Molecular-biological characterization of marek’s disease virus. I. Identification of virus-specific polypeptides in infected cells. Virology 1982, 121, 116–132. [Google Scholar] [CrossRef]
- OIE. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. 2015. Available online: http://www.oie.int/standard-setting/terrestrial-manual/access-online/ (accessed on 25 April 2019).
- Peeters, B.; de Wind, N.; Hooisma, M.; Wagenaar, F.; Gielkens, A.; Moormann, R. Pseudorabies virus envelope glycoproteins gp50 and gii are essential for virus penetration, but only gii is involved in membrane fusion. J. Virol. 1992, 66, 894–905. [Google Scholar] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Amer. J. Trop. Med. Hygiene 1938, 27, 493–497. [Google Scholar]
- Atienza, J.M.; Yu, N.; Kirstein, S.L.; Xi, B.; Wang, X.; Xu, X.; Abassi, Y.A. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev. Technol. 2006, 4, 597–607. [Google Scholar] [CrossRef]
- Poen, M.J.; Bestebroer, T.M.; Vuong, O.; Scheuer, R.D.; van der Jeugd, H.P.; Kleyheeg, E.; Eggink, D.; Lexmond, P.; van den Brand, J.M.A.; Begeman, L.; et al. Local amplification of highly pathogenic avian influenza h5n8 viruses in wild birds in the Netherlands, 2016 to 2017. Euro. Surveill. 2018, 23, 17-00449. [Google Scholar] [CrossRef]
- Kleyheeg, E.; Slaterus, R.; Bodewes, R.; Rijks, J.M.; Spierenburg, M.A.H.; Beerens, N.; Kelder, L.; Poen, M.J.; Stegeman, J.A.; Fouchier, R.A.M.; et al. Deaths among wild birds during highly pathogenic avian influenza a (h5n8) virus outbreak, the Netherlands. Emerg. Infect. Dis. 2017, 23, 2050–2054. [Google Scholar] [CrossRef]
- Globig, A.; Staubach, C.; Sauter-Louis, C.; Dietze, K.; Homeier-Bachmann, T.; Probst, C.; Gethmann, J.; Depner, K.R.; Grund, C.; Harder, T.C.; et al. Highly pathogenic avian influenza h5n8 clade 2.3.4.4b in germany in 2016/2017. Front. Vet. Sci. 2017, 4, 240. [Google Scholar] [CrossRef] [PubMed]
- Spackman, E.; Pantin-Jackwood, M.J.; Kapczynski, D.R.; Swayne, D.E.; Suarez, D.L. H5N2 highly pathogenic avian influenza viruses from the us 2014–2015 outbreak have an unusually long pre-clinical period in turkeys. BMC Vet. Res. 2016, 12, 260. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| H5N5 Genotype | H5N5 Isolate Number | Host | Collection Date | Collection Location |
|---|---|---|---|---|
| H5N5-GT1 | 1 | Tufted duck | 2016-11-14 | Netherlands |
| H5N5-GT2 | 2 | Environment | 2016-10-01 | Russia |
| 3 | Swan | 2016-12-13 | Germany | |
| 4 | Mute swan | 2016-12-13 | Netherlands | |
| 5 a | Barnacle goose | 2016-12-22 | Germany | |
| 6 | Greylag goose | 2016-12-27 | Germany | |
| 7 | Mute swan | 2016-12-27 | Croatia | |
| 8 | Eurasian wigeon | 2016-12-29 | Italy | |
| 9 | Gadwall | 2017-01-10 | Italy | |
| 10 | Mute swan | 2017-01-20 | Croatia | |
| 11 | Grey heron | 2017-01-22 | Germany | |
| 12 a | Mute swan | 2017-01-31 | Poland | |
| 13 | Common buzzard | 2017-02-06 | Germany | |
| 14 | Mute swan | 2017-02-14 | Hungary | |
| 15 | Chicken | 2017-03-07 | Croatia | |
| H5N5-GT3 | 16 | Turkey | 2017-01-22 | Germany |
| 17 | Cormorant | 2017-01-30 | Germany | |
| 18 | Egret | 2017-02-14 | Germany | |
| 19 | Goose | 2017-05-22 | Netherlands | |
| Unknown | 20 a | Mute swan | 2017-02-09 | Czech Republic |
| 21 a | Spot-billed pelican | 2017-02-14 | Czech Republic |
| H5N5-GT2 Viruses (Node 1) | European H5N5-GT2 Viruses (Node 2) | H5N5-GT3 Viruses (Node 3) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Segment | Median TMRCA | Lower 95% HPD | Upper 95% HPD | Posterior | Median TMRCA | Lower 95% HPD | Upper 95% HPD | Posterior | Median TMRCA | Lower 95% HPD | Upper 95% HPD | Posterior |
| PB2 | Mar-2016 | Aug-2015 | Aug-2016 | 0.9997 | Sep-2016 | May-2016 | Nov-2016 | 0.9997 | Oct-2016 | Aug-2016 | Nov-2016 | 0.4823 |
| PB1 | Dec-2015 | Apr-2015 | Jun-2016 | 0.9983 | Jul-2016 | Feb-2016 | Oct-2016 | 0.9982 | Oct-2016 | May-2016 | Jan-2017 | 0.9971 |
| PA | Aug-2016 | Jun-2016 | Sep-2016 | 0.9982 | Oct-2016 | Sep-2016 | Nov-2016 | 0.9995 | Nov-2016 | Oct-2016 | Dec-2016 | 0.0942 |
| HA | Jun-2016 | Jan-2016 | Aug-2016 | 0.9995 | Oct-2016 | Oct-2016 | Jun-2016 | 0.9991 | Dec-2016 | Oct-2016 | Jan-2017 | 0.9991 |
| NP | Mar-2016 | Aug-2015 | Aug-2016 | 0.9988 | Aug-2016 | Apr-2016 | Aug-2016 | 0.9988 | Sep-2016 | Mar-2016 | Dec-2016 | 0.9991 |
| NA | Nov-2015 | Apr-2015 | May-2016 | 0.9997 | Apr-2016 | Nov-2015 | Aug-2016 | 0.9929 | Oct-2016 | Jun-2016 | Jan-2017 | 0.9713 |
| MP | Aug-2016 | Jun-2016 | Sep-2016 | 0.9929 | Nov-2016 | Nov-2016 | Dec-2016 | 0.9928 | Jan-2017 | Dec-2016 | Jan-2017 | 0.0325 |
| NS | Jul-2016 | Mar-2016 | Sep-2016 | 0.9992 | Nov-2016 | Jun-2016 | Aug-2016 | 0.9835 | Dec-2016 | Nov-2016 | Dec-2016 | 0.0038 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergervoet, S.A.; Ho, C.K.Y.; Heutink, R.; Bossers, A.; Beerens, N. Spread of Highly Pathogenic Avian Influenza (HPAI) H5N5 Viruses in Europe in 2016–2017 Appears Related to the Timing of Reassortment Events. Viruses 2019, 11, 501. https://doi.org/10.3390/v11060501
Bergervoet SA, Ho CKY, Heutink R, Bossers A, Beerens N. Spread of Highly Pathogenic Avian Influenza (HPAI) H5N5 Viruses in Europe in 2016–2017 Appears Related to the Timing of Reassortment Events. Viruses. 2019; 11(6):501. https://doi.org/10.3390/v11060501
Chicago/Turabian StyleBergervoet, Saskia A., Cynthia K. Y. Ho, Rene Heutink, Alex Bossers, and Nancy Beerens. 2019. "Spread of Highly Pathogenic Avian Influenza (HPAI) H5N5 Viruses in Europe in 2016–2017 Appears Related to the Timing of Reassortment Events" Viruses 11, no. 6: 501. https://doi.org/10.3390/v11060501
APA StyleBergervoet, S. A., Ho, C. K. Y., Heutink, R., Bossers, A., & Beerens, N. (2019). Spread of Highly Pathogenic Avian Influenza (HPAI) H5N5 Viruses in Europe in 2016–2017 Appears Related to the Timing of Reassortment Events. Viruses, 11(6), 501. https://doi.org/10.3390/v11060501