Establishment of a Cell Culture Model of Persistent Flaviviral Infection: Usutu Virus Shows Sustained Replication during Passages and Resistance to Extinction by Antiviral Nucleosides


Abstract
:1. Introduction
2. Materials and Methods
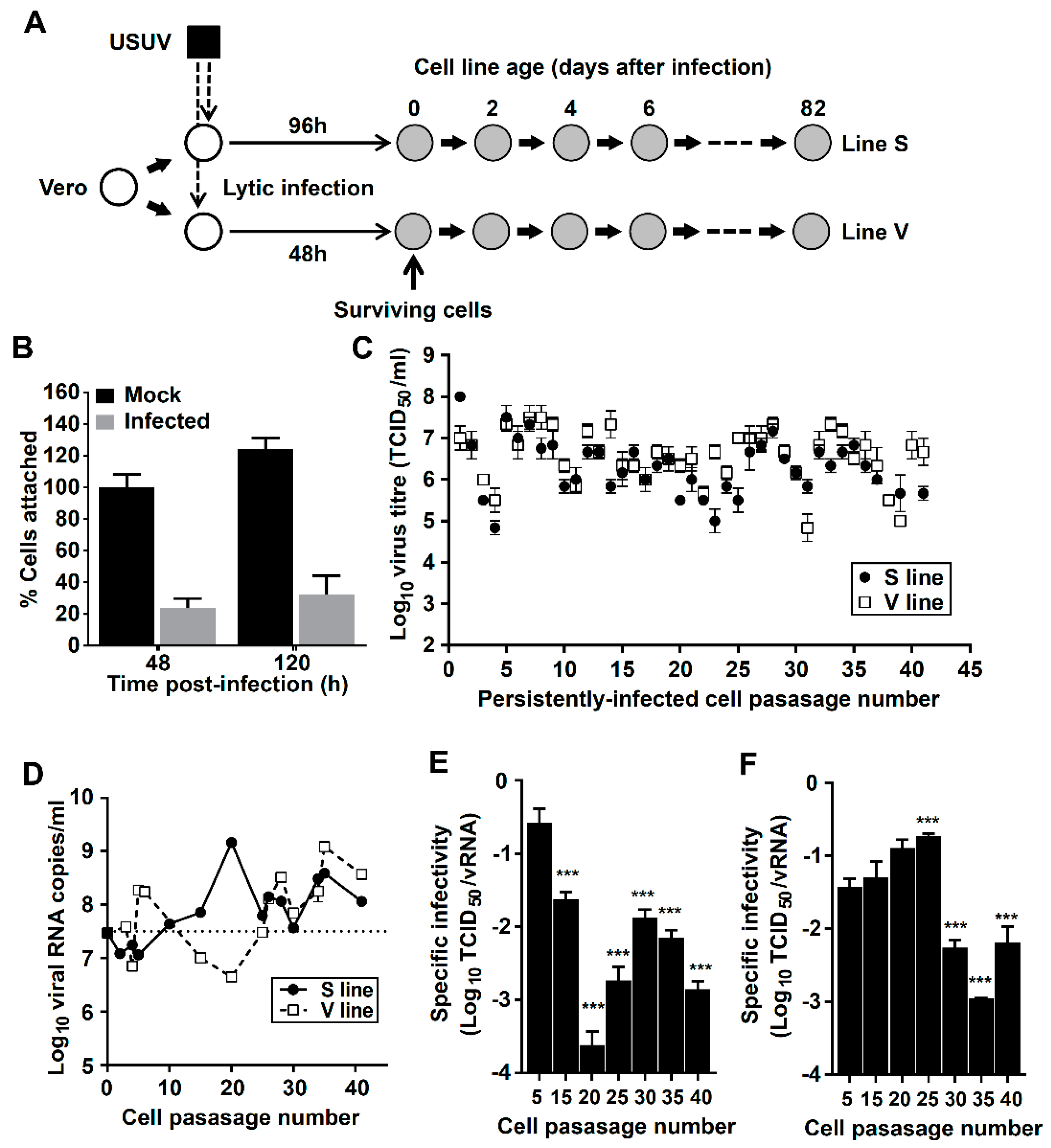
2.1. Cells, Virus, and Establishment of Persistently Infected Cultures
2.2. Virus Titration
2.3. Treatment with Antiviral Compounds
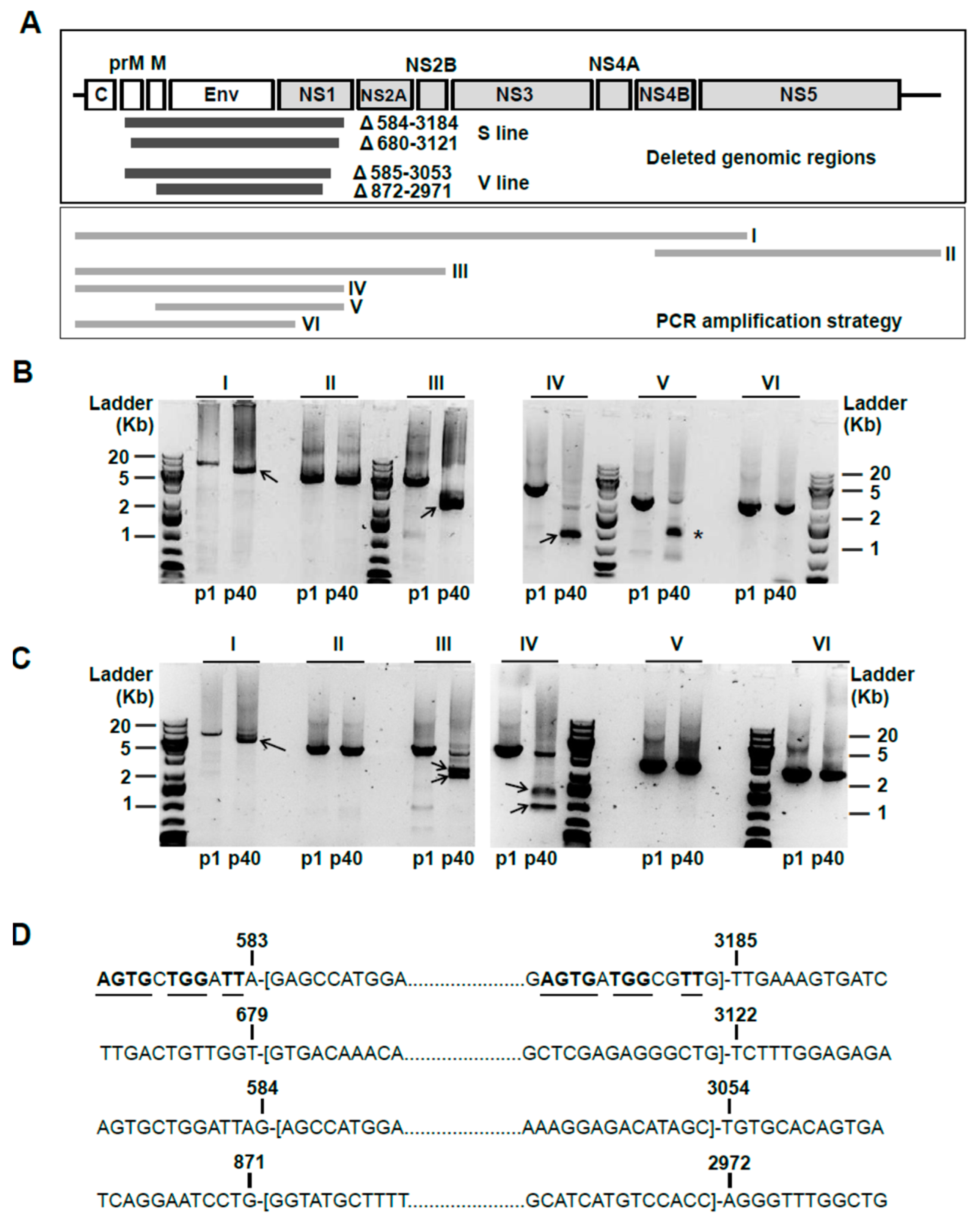
2.4. Viral RNA Extraction, RT-PCR Amplification, and Sequence Analysis
2.5. Quantitative PCR Analysis of Virus Populations
2.6. Statistical Analysis
3. Results
3.1. Establishment of Cell Lines Persistently Infected with USUV
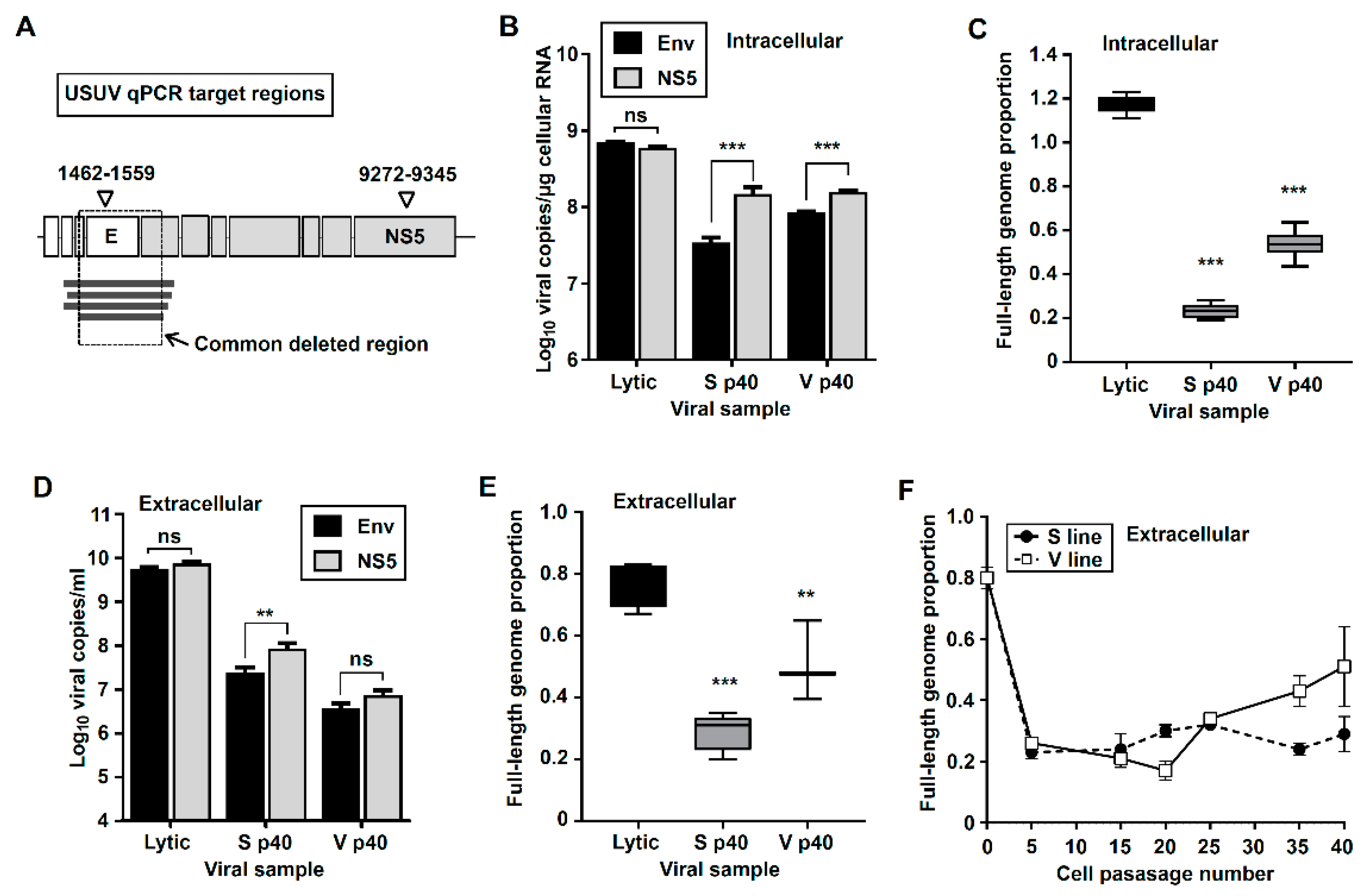
3.2. Quantification of Truncated and Standard Viral Genomes in Persistently-Infected Cells
3.3. Non-Synonymous Mutations are Accumulated in USUV Proteins NS4A and NS4B
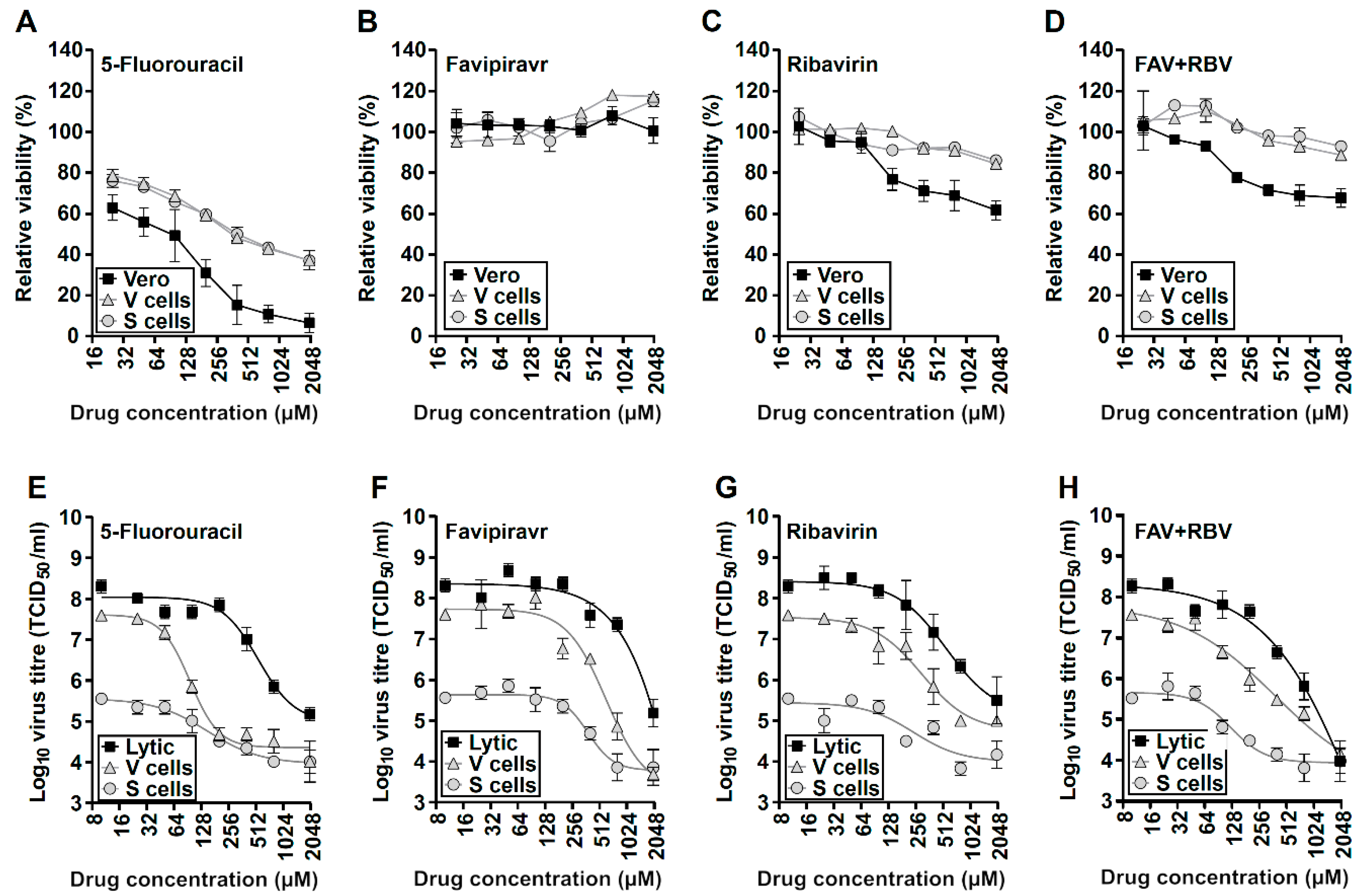
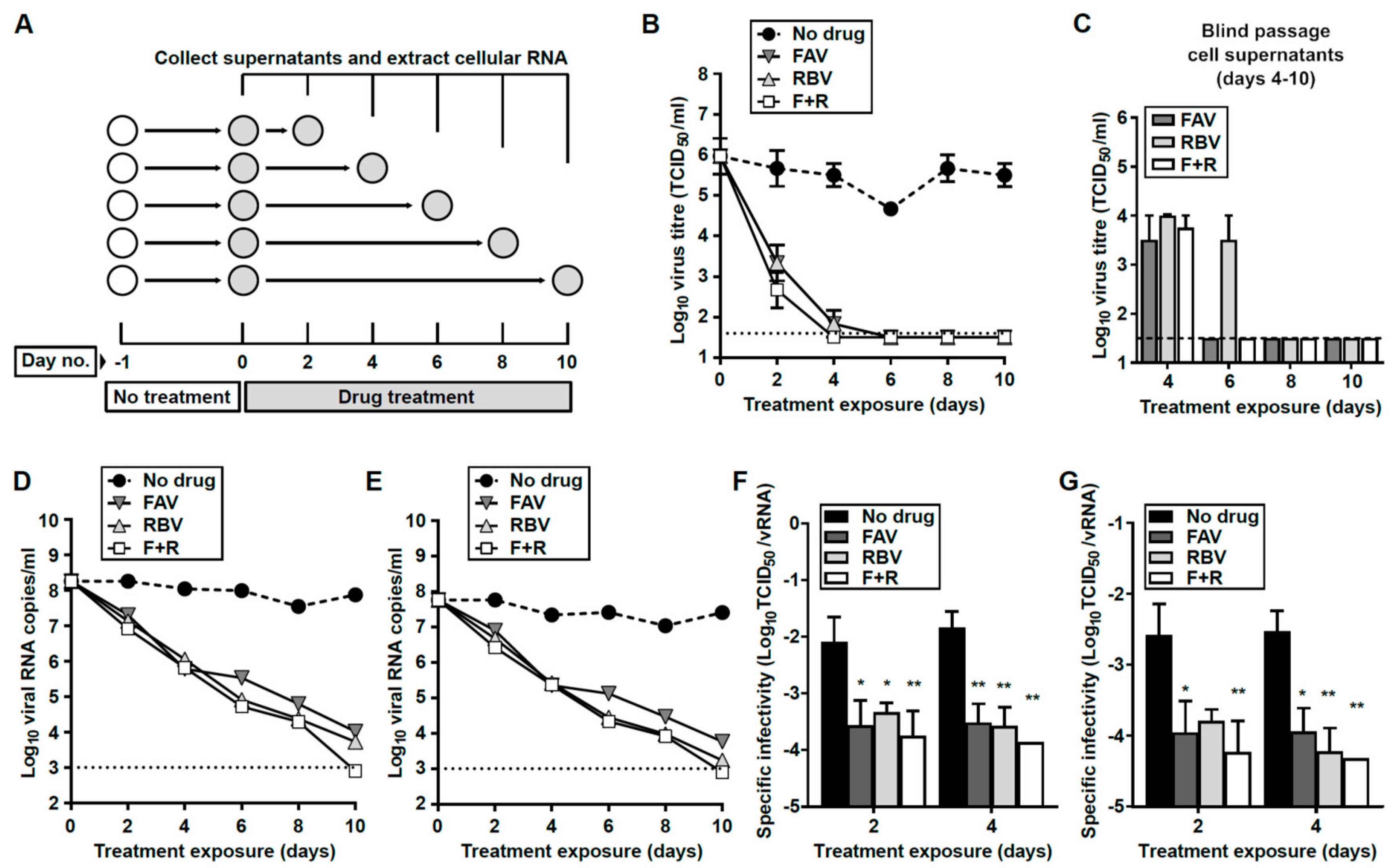
3.4. Broad-Range Nucleoside Drugs Strongly Inhibit Persistent USUV
3.5. Prolonged Treatment with Nucleoside Drugs Abolishes Infectivity in Persistently-Infected Cell Supernatants
3.6. Interruption of Drug Treatment Results in the Recovery of Viral Infectivity
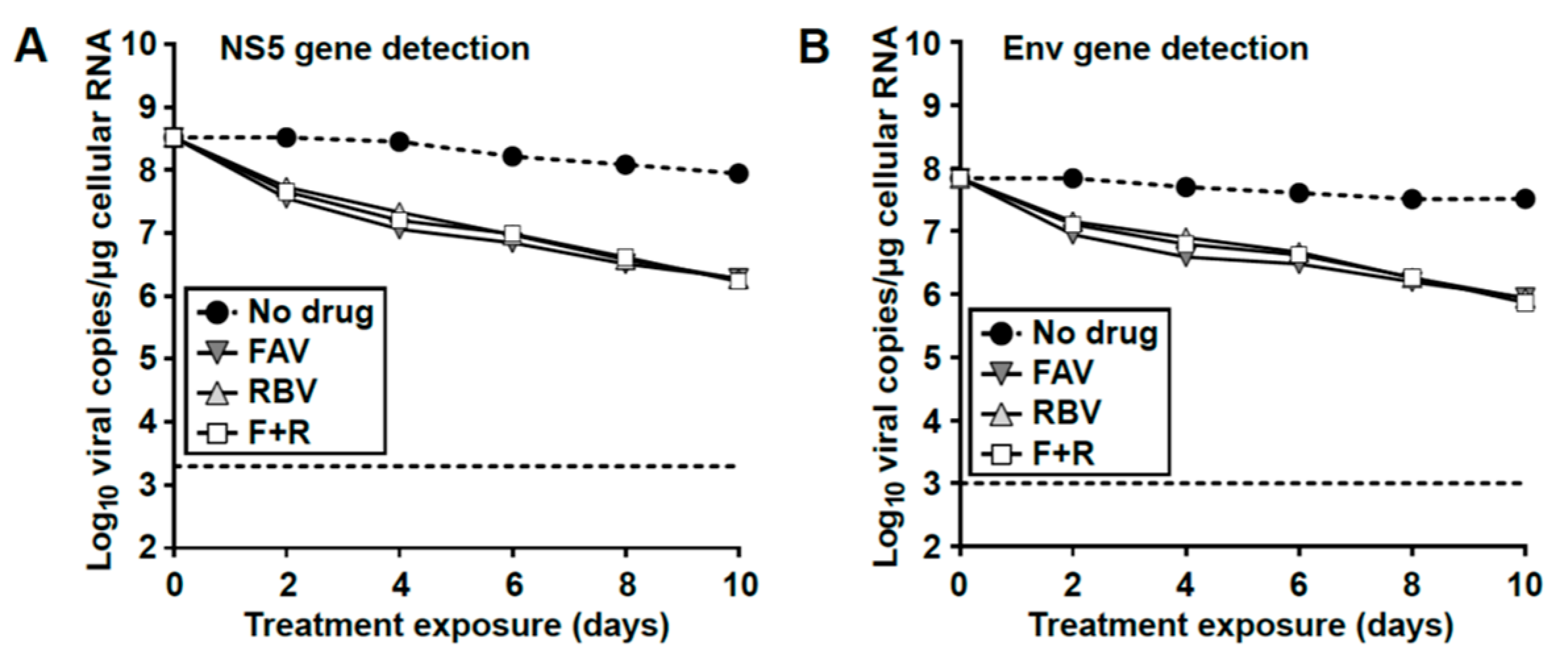
3.7. Loss of Extracellular Infectivity is not Accompanied by Intracellular Extinction of USUV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef]
- Murali, A.R.; Kotwal, V.; Chawla, S. Chronic hepatitis E: A brief review. World J. Hepatol. 2015, 7, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Elbedewy, T.A.; El-Serafy, M.; El-Toukhy, N.; Ahmed, W.; El Din, Z.A. Hepatitis C virus: A global view. World J. Hepatol. 2015, 7, 2676–2680. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.; Gkrania-Klotsas, E.; Kumararatne, D. Chronic norovirus infection and common variable immunodeficiency. Clin. Exp. Immunol. 2017, 188, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Capizzi, T.; Makari-Judson, G.; Steingart, R.; Mertens, W.C. Chronic diarrhea associated with persistent norovirus excretion in patients with chronic lymphocytic leukemia: Report of two cases. BMC Infect. Dis. 2011, 11, 131. [Google Scholar] [CrossRef]
- Mlera, L.; Melik, W.; Bloom, M.E. The role of viral persistence in flavivirus biology. Pathog. Dis. 2014, 71, 137–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzoni, T.B.; López, C.B. Defective (interfering) viral genomes re-explored: Impact on antiviral immunity and virus persistence. Future Virol. 2018, 13, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.G.; Granich, R. Ending AIDS: Myth or reality? Lancet 2017, 390, 357. [Google Scholar] [CrossRef]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Hoarau, J.J.; Jaffar Bandjee, M.C.; Krejbich Trotot, P.; Das, T.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent Chronic Inflammation and Infection by Chikungunya Arthritogenic Alphavirus in Spite of a Robust Host Immune Response. J. Immunol. 2010, 184, 5914–5927. [Google Scholar] [CrossRef] [Green Version]
- Gould, E.A.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Garcia, M.N.; Hasbun, R.; Murray, K.O. Persistence of West Nile virus. Microbes Infect. 2015, 17, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.O.; Kolodziej, S.; Ronca, S.E.; Gorchakov, R.; Navarro, P.; Nolan, M.S.; Podoll, A.; Finkel, K.; Mandayam, S. Visualization of West Nile Virus in Urine Sediment using Electron Microscopy and Immunogold up to Nine Years Postinfection. Am. J. Trop. Med. Hyg. 2017, 97, 1913–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, V.; Xie, G.; Li, B.; Farris, T.; Welte, T.; Gong, B.; Boor, P.; Wu, P.; Tang, S.-J.; Tesh, R.; et al. A Hamster-Derived West Nile Virus Isolate Induces Persistent Renal Infection in Mice. PLoS Negl. Trop. Dis. 2013, 7, e2275. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.P.; Travassos da Rosa, A.P.A.; Nunes, M.R.; Xiao, S.-Y.; Tesh, R.B. Pathogenesis of Modoc Virus (Flaviviridae; Flavivirus) in Persistently Infected Hamsters. Am. J. Trop. Med. Hyg. 2013, 88, 455–460. [Google Scholar] [CrossRef]
- Davis, J.W.; Hardy, J.L.; Reeves, W.C. Modoc viral infections in the deer mouse Peromyscus maniculatus. Infect. Immun. 1974, 10, 1362–1369. [Google Scholar] [PubMed]
- Brinton, M.A. Isolation of a replication-efficient mutant of West Nile virus from a persistently infected genetically resistant mouse cell culture. J. Virol. 1981, 39, 413–421. [Google Scholar] [Green Version]
- Prisant, N.; Bujan, L.; Benichou, H.; Hayot, P.-H.; Pavili, L.; Lurel, S.; Herrmann, C.; Janky, E.; Joguet, G. Zika virus in the female genital tract. Lancet Infect. Dis. 2016, 16, 1000–1001. [Google Scholar] [CrossRef] [Green Version]
- Paz-Bailey, G.; Rosenberg, E.S.; Doyle, K.; Munoz-Jordan, J.; Santiago, G.A.; Klein, L.; Perez-Padilla, J.; Medina, F.A.; Waterman, S.H.; Gubern, C.G.; et al. Persistence of Zika Virus in Body Fluids—Preliminary Report. N. Engl. J. Med. 2017, 379, NEJMoa1613108. [Google Scholar]
- D’Ortenzio, E.; Matheron, S.; de Lamballerie, X.; Hubert, B.; Piorkowski, G.; Maquart, M.; Descamps, D.; Damond, F.; Yazdanpanah, Y.; Leparc-Goffart, I. Evidence of Sexual Transmission of Zika Virus. N. Engl. J. Med. 2016, 374, 2195–2198. [Google Scholar] [CrossRef]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Gritsun, T.S.; Frolova, T.V.; Zhankov, A.I.; Armesto, M.; Turner, S.L.; Frolova, M.P.; Pogodina, V.V.; Lashkevich, V.A.; Gould, E.A. Characterization of a Siberian Virus Isolated from a Patient with Progressive Chronic Tick-Borne Encephalitis. J. Virol. 2003, 77, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, V.; Desai, A.S.; Shenoy, P.K.; Satishchandra, P.; Chandramuki, A.; Gourie-Devi, M. Persistence of Japanese encephalitis virus in the human nervous system. J. Med. Virol. 1993, 40, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.L.; Anderson, S.L.; Lord, C.C.; Smartt, C.T.; Tabachnick, W.J. Relationships between infection, dissemination, and transmission of West Nile virus RNA in Culex pipiens quinquefasciatus (Diptera: Culicidae). J. Med. Entomol. 2012, 49, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Girard, Y.A.; Klingler, K.A.; Higgs, S. West Nile Virus Dissemination and Tissue Tropisms in Orally Infected Culex pipiens quinquefasciatus. Vector-Borne Zoonotic Dis. 2004, 4, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Belova, O.A.; Litov, A.G.; Kholodilov, I.S.; Kozlovskaya, L.I.; Bell-Sakyi, L.; Romanova, L.I.; Karganova, G.G. Properties of the tick-borne encephalitis virus population during persistent infection of ixodid ticks and tick cell lines. Ticks Tick-Borne Dis. 2017, 8, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Mlera, L.; Offerdahl, D.K.; Martens, C.; Porcella, S.F.; Melik, W.; Bloom, M.E. Development of a Model System for Tick-Borne Flavivirus Persistence in HEK 293T Cells. MBio 2015, 6, e00614-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlera, L.; Lam, J.; Offerdahl, D.K.; Martens, C.; Sturdevant, D.; Turner, C.V.; Porcella, S.F.; Bloom, M.E. Transcriptome Analysis Reveals a Signature Profile for Tick-Borne Flavivirus Persistence in HEK 293T Cells. MBio 2016, 7, e00314-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A Three-Dimensional Comparison of Tick-Borne Flavivirus Infection in Mammalian and Tick Cell Lines. PLoS ONE 2012, 7, e47912. [Google Scholar] [CrossRef] [PubMed]
- Mlera, L.; Melik, W.; Offerdahl, D.; Dahlstrom, E.; Porcella, S.; Bloom, M. Analysis of the Langat Virus Genome in Persistent Infection of an Ixodes scapularis Cell Line. Viruses 2016, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, R.; Chandra, P.K.; Ferraris, P.; Kurt, R.; Song, K.; Garry, R.F.; Reiss, K.; Coe, I.R.; Furihata, T.; Balart, L.A.; et al. Persistent Hepatitis C Virus Infection Impairs Ribavirin Antiviral Activity through Clathrin-Mediated Trafficking of Equilibrative Nucleoside Transporter 1. J. Virol. 2015, 89, 626–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, V.; Ávila-Pérez, G.; Mingorance, L.; Gastaminza, P. A Cell Culture Model for Persistent HCV Infection. In Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2019; Volume 1911, pp. 157–168. [Google Scholar]
- Tsai, P.; Lin, C.C.; Sun, H.Y.; Lee, J.C.; Chang, T.T.; Young, K.C. Viral dynamics of persistent hepatitis C virus infection in high-sensitive reporter cells resemble patient’s viremia. J. Microbiol. Immunol. Infect. 2017, 51, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Chung, J.; Stamataki, Z.; Isogawa, M.; Cheng, G.; McKeating, J.A.; Chisari, F.V. Persistent hepatitis C virus infection in vitro: Coevolution of virus and host. J. Virol. 2006, 80, 11082–11093. [Google Scholar] [CrossRef] [PubMed]
- Rijks, J.M.; Kik, M.L.; Slaterus, R.; Foppen, R.; Stroo, A.; IJzer, J.; Stahl, J.; Gröne, A.; Koopmans, M.; van der Jeugd, H.P.; et al. Widespread Usutu virus outbreak in birds in The Netherlands, 2016. Eurosurveillance 2016, 21, 30391. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, U.; Ye, J.; Ruan, X.; Wan, S.; Zhu, B.; Cao, S. Usutu virus: An emerging flavivirus in Europe. Viruses 2015, 7, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Jungbauer, C.; Aberle, S.W.; Kolodziejek, J.; Dimmel, K.; Stiasny, K.; Allerberger, F.; Nowotny, N. Usutu virus infections among blood donors, Austria, July and August 2017—Raising awareness for diagnostic challenges. Eurosurveillance 2017, 22, 17–00644. [Google Scholar] [CrossRef] [PubMed]
- Aberle, S.W.; Kolodziejek, J.; Jungbauer, C.; Stiasny, K.; Aberle, J.H.; Zoufaly, A.; Hourfar, M.K.; Weidner, L.; Nowotny, N. Increase in human West Nile and Usutu virus infections, Austria, 2018. Eurosurveillance 2018, 23, 1800545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadar, D.; Maier, P.; Müller, S.; Kress, J.; Chudy, M.; Bialonski, A.; Schlaphof, A.; Jansen, S.; Jöst, H.; Tannich, E.; et al. Blood donor screening for West Nile virus (WNV) revealed acute Usutu virus (USUV) infection, Germany, September 2016. Eurosurveillance 2017, 22, 30501. [Google Scholar] [CrossRef]
- Grottola, A.; Marcacci, M.; Tagliazucchi, S.; Gennari, W.; Di Gennaro, A.; Orsini, M.; Monaco, F.; Marchegiano, P.; Marini, V.; Meacci, M.; et al. Usutu virus infections in humans: A retrospective analysis in the municipality of Modena, Italy. Clin. Microbiol. Infect. 2017, 23, 33–37. [Google Scholar] [CrossRef]
- Santini, M.; Vilibic-Cavlek, T.; Barsic, B.; Barbic, L.; Savic, V.; Stevanovic, V.; Listes, E.; Di Gennaro, A.; Savini, G. First cases of human Usutu virus neuroinvasive infection in Croatia, August–September 2013: Clinical and laboratory features. J. Neurovirol. 2015, 21, 92–97. [Google Scholar] [CrossRef]
- Pecorari, M.; Longo, G.; Gennari, W.; Grottola, A.; Sabbatini, A.; Tagliazucchi, S.; Savini, G.; Monaco, F.; Simone, M.; Lelli, R.; et al. First human case of Usutu virus neuroinvasive infection, Italy, August–September 2009. Eurosurveillance 2009, 14, 19446. [Google Scholar] [PubMed]
- Simonin, Y.; Sillam, O.; Carles, M.J.; Gutierrez, S.; Gil, P.; Constant, O.; Martin, M.F.; Grard, G.; Van de Perre, P.; Salinas, S.; et al. Human Usutu Virus Infection with Atypical Neurologic Presentation, Montpellier, France, 2016. Emerg. Infect. Dis. 2018, 24, 875–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetro, J.A. Is Usutu virus ready for prime time? Microbes Infect. 2017, 19, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.R.; Sempere, R.N.; Meyn, P.; Polacek, C.; Arias, A. Extinction of Zika virus and Usutu virus by lethal mutagenesis reveals different patterns of sensitivity to three mutagenic drugs. Antimicrob. Agents Chemother. 2018, 62, e00380-18. [Google Scholar] [CrossRef] [PubMed]
- Segura Guerrero, N.A.; Sharma, S.; Neyts, J.; Kaptein, S.J.F. Favipiravir inhibits in vitro Usutu virus replication and delays disease progression in an infection model in mice. Antivir. Res. 2018, 160, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Weissenböck, H.; Kolodziejek, J.; Url, A.; Lussy, H.; Rebel-Bauder, B.; Nowotny, N. Emergence of Usutu virus, an African Mosquito-Borne Flavivirus of the Japanese Encephalitis Virus Group, Central Europe. Emerg. Infect. Dis. 2002, 8, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D. Measuring HCV Infectivity Produced in Cell Culture and In Vivo. In Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2019; Volume 510, pp. 329–336. [Google Scholar]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Cavrini, F.; Pepa, M.E.D.; Gaibani, P.; Pierro, A.M.; Rossini, G.; Landini, M.P.; Sambri, V. A rapid and specific real-time RT-PCR assay to identify Usutu virus in human plasma, serum, and cerebrospinal fluid. J. Clin. Virol. 2011, 50, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Agudo, R.; Tejero, H.; Manrubia, S.C.; Domingo, E. Potential benefits of sequential inhibitor-mutagen treatments of RNA virus infections. PLoS Pathog. 2009, 5, e1000658. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Romero, E.; De Oya, N.J.; Domingo, E.; Saiza, J.C. Extinction of west nile virus by favipiravir through lethal mutagenesis. Antimicrob. Agents Chemother. 2017, 61, e01400-17. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Bocan, T.M.; Wells, J.; Wetzel, K.S.; Van Tongeren, S.A.; Dong, L.; Garza, N.L.; Donnelly, G.; Cazares, L.H.; Nuss, J.; et al. Efficacy of favipiravir (T-705) in nonhuman primates infected with Ebola virus or Marburg virus. Antivir. Res. 2018, 151, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kaaijk, P.; Luytjes, W. Are we prepared for emerging flaviviruses in Europe? Challenges for vaccination. Hum. Vaccin. Immunother. 2018, 14, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Daep, C.A.; Muñoz-Jordán, J.L.; Eugenin, E.A. Flaviviruses, an expanding threat in public health: Focus on dengue, West Nile, and Japanese encephalitis virus. J. Neurovirol. 2014, 20, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.-S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.-H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef]
- Bielefeldt-Ohmann, H.; Bosco-Lauth, A.; Hartwig, A.-E.; Uddin, M.J.; Barcelon, J.; Suen, W.W.; Wang, W.; Hall, R.A.; Bowen, R.A. Characterization of non-lethal West Nile Virus (WNV) infection in horses: Subclinical pathology and innate immune response. Microb. Pathog. 2017, 103, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, A.-B.; Escribano-Romero, E.; Martín-Acebes, M.A.; Petrovic, T.; Saiz, J.-C. Limited susceptibility of mice to Usutu virus (USUV) infection and induction of flavivirus cross-protective immunity. Virology 2015, 482, 67–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chvala, S.; Bakonyi, T.; Hackl, R.; Hess, M.; Nowotny, N.; Weissenböck, H. Limited pathogenicity of Usutu virus for the domestic chicken (Gallus domesticus). Avian Pathol. 2005, 34, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Martínez, A.B.; Vega-Almeida, T.O.; Salas-Benito, M.; García-Espitia, M.; De Nova-Ocampo, M.; del Ángel, R.M.; Salas-Benito, J.S. Detection and sequencing of defective viral genomes in C6/36 cells persistently infected with dengue virus 2. Arch. Virol. 2013, 158, 583–599. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.U.; Hodgetts, S.I.; Mackenzie, J.S.; Urosevic, N. Characterization of defective viral RNA produced during persistent infection of Vero cells with Murray Valley encephalitis virus. J. Virol. 1998, 72, 2474–2482. [Google Scholar] [PubMed]
- Pesko, K.N.; Fitzpatrick, K.A.; Ryan, E.M.; Shi, P.-Y.; Zhang, B.; Lennon, N.J.; Newman, R.M.; Henn, M.R.; Ebel, G.D. Internally deleted WNV genomes isolated from exotic birds in New Mexico: Function in cells, mosquitoes, and mice. Virology 2012, 427, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.; Zhang, B.; Lim, P.-Y.; Yuan, Z.; Bernard, K.A.; Shi, P.-Y. Exclusion of West Nile virus superinfection through RNA replication. J. Virol. 2009, 83, 11765–11776. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Lühken, R.; van der Jeugd, H.; Garigliany, M.; Ziegler, U.; Keller, M.; Lahoreau, J.; Lachmann, L.; Becker, N.; Kik, M.; et al. Widespread activity of multiple lineages of Usutu virus, western Europe, 2016. Eurosurveillance 2017, 22, 30452. [Google Scholar] [CrossRef] [PubMed]
- Sissoko, D.; Laouenan, C.; Folkesson, E.; M’Lebing, A.-B.; Beavogui, A.-H.; Baize, S.; Camara, A.-M.; Maes, P.; Shepherd, S.; Danel, C.; et al. Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea. PLoS Med. 2016, 13, e1001967. [Google Scholar] [CrossRef] [PubMed]
- Thorne, L.; Arias, A.; Goodfellow, I. Advances Toward a Norovirus Antiviral: From Classical Inhibitors to Lethal Mutagenesis. J. Infect. Dis. 2016, 213 (Suppl. 1), S27–S31. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Domingo, E. Antiviral Strategies Based on Lethal Mutagenesis and Error Threshold. Curr. Top. Microbiol. Immunol. 2016, 392, 323–339. [Google Scholar] [PubMed]
- Arias, A.; Thorne, L.; Goodfellow, I. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. Elife 2014, 3, e03679. [Google Scholar] [CrossRef]
- Ruis, C.; Brown, L.-A.K.; Roy, S.; Atkinson, C.; Williams, R.; Burns, S.O.; Yara-Romero, E.; Jacobs, M.; Goldstein, R.; Breuer, J.; et al. Mutagenesis in Norovirus in Response to Favipiravir Treatment. N. Engl. J. Med. 2018, 379, 2173–2176. [Google Scholar] [CrossRef] [Green Version]
- Dietz, J.; Schelhorn, S.-E.; Fitting, D.; Mihm, U.; Susser, S.; Welker, M.-W.; Füller, C.; Däumer, M.; Teuber, G.; Wedemeyer, H.; et al. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. J. Virol. 2013, 87, 6172–6181. [Google Scholar] [CrossRef]
- Zou, J.; Xie, X.; Wang, Q.-Y.; Dong, H.; Lee, M.Y.; Kang, C.; Yuan, Z.; Shi, P.-Y. Characterization of dengue virus NS4A and NS4B protein interaction. J. Virol. 2015, 89, 3455–3470. [Google Scholar] [CrossRef]
- Blázquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martín-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, A.-B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.-C.; Martín-Acebes, M.A. Infection with Usutu Virus Induces an Autophagic Response in Mammalian Cells. PLoS Negl. Trop. Dis. 2013, 7, e2509. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Position | USUV Parental Stock | S p40 | V p40 | Viral Gene | Amino Acid Replacement |
|---|---|---|---|---|---|
| G775 * | A | A | A | M | E9K |
| C891 1 | U | U | M | − | |
| C1815 1 | U | U | Env | − | |
| C3661 | U | NS2A | − | ||
| A3867 | G | NS2A | − | ||
| C4958 | U | NS3 | T118M | ||
| C5432 * | U | U | U | NS3 | A276V |
| G6528 | U | NS4A | E22D | ||
| A6532 2 | C | NS4A | F24L | ||
| U6534 2 | A | NS4A | F24L | ||
| G6595 1 | C | C | NS4A | E45N | |
| U7175 | G | NS4B | L89R | ||
| C7184 1 | U | U | NS4B | T92M | |
| U7197 * | G | G | G | NS4B | F96L |
| U7288 | C | NS4B | Y127H | ||
| U7424 | G | NS4B | M172R | ||
| A10311 * | G | G | G | NS5 | − |
| Cell Line | Compound | IC50 (µM) | CC50 (µM) | Selectivity Index |
|---|---|---|---|---|
| Vero cells | 5-Fluorouracil | 71 | 66 | 1.1 |
| Favipiravir | 315 | >2000 | >6.3 | |
| Ribavirin | 163 | >2000 | >12.3 | |
| Favipiravir + Ribavirin | 60 * | >2000 * | >33.3 | |
| V p39 | 5-Fluorouracil | 42 | 467 | 11.1 |
| Favipiravir | 197 | >2000 | >10.2 | |
| Ribavirin | 71 | >2000 | >28.2 | |
| Favipiravir + Ribavirin | 36 * | >2000 * | >55.6 | |
| S p39 | 5-Fluorouracil | 56 | 472 | 8.4 |
| Favipiravir | 241 | >2000 | >8.3 | |
| Ribavirin | 80 | >2000 | >25.0 | |
| Favipiravir + Ribavirin | 78 * | >2000 * | >25.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sempere, R.N.; Arias, A. Establishment of a Cell Culture Model of Persistent Flaviviral Infection: Usutu Virus Shows Sustained Replication during Passages and Resistance to Extinction by Antiviral Nucleosides. Viruses 2019, 11, 560. https://doi.org/10.3390/v11060560
Sempere RN, Arias A. Establishment of a Cell Culture Model of Persistent Flaviviral Infection: Usutu Virus Shows Sustained Replication during Passages and Resistance to Extinction by Antiviral Nucleosides. Viruses. 2019; 11(6):560. https://doi.org/10.3390/v11060560
Chicago/Turabian StyleSempere, Raquel Navarro, and Armando Arias. 2019. "Establishment of a Cell Culture Model of Persistent Flaviviral Infection: Usutu Virus Shows Sustained Replication during Passages and Resistance to Extinction by Antiviral Nucleosides" Viruses 11, no. 6: 560. https://doi.org/10.3390/v11060560
APA StyleSempere, R. N., & Arias, A. (2019). Establishment of a Cell Culture Model of Persistent Flaviviral Infection: Usutu Virus Shows Sustained Replication during Passages and Resistance to Extinction by Antiviral Nucleosides. Viruses, 11(6), 560. https://doi.org/10.3390/v11060560