Modulation of Innate Immune Signaling Pathways by Herpesviruses

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Modulation of Inflammatory Response by Herpesviruses
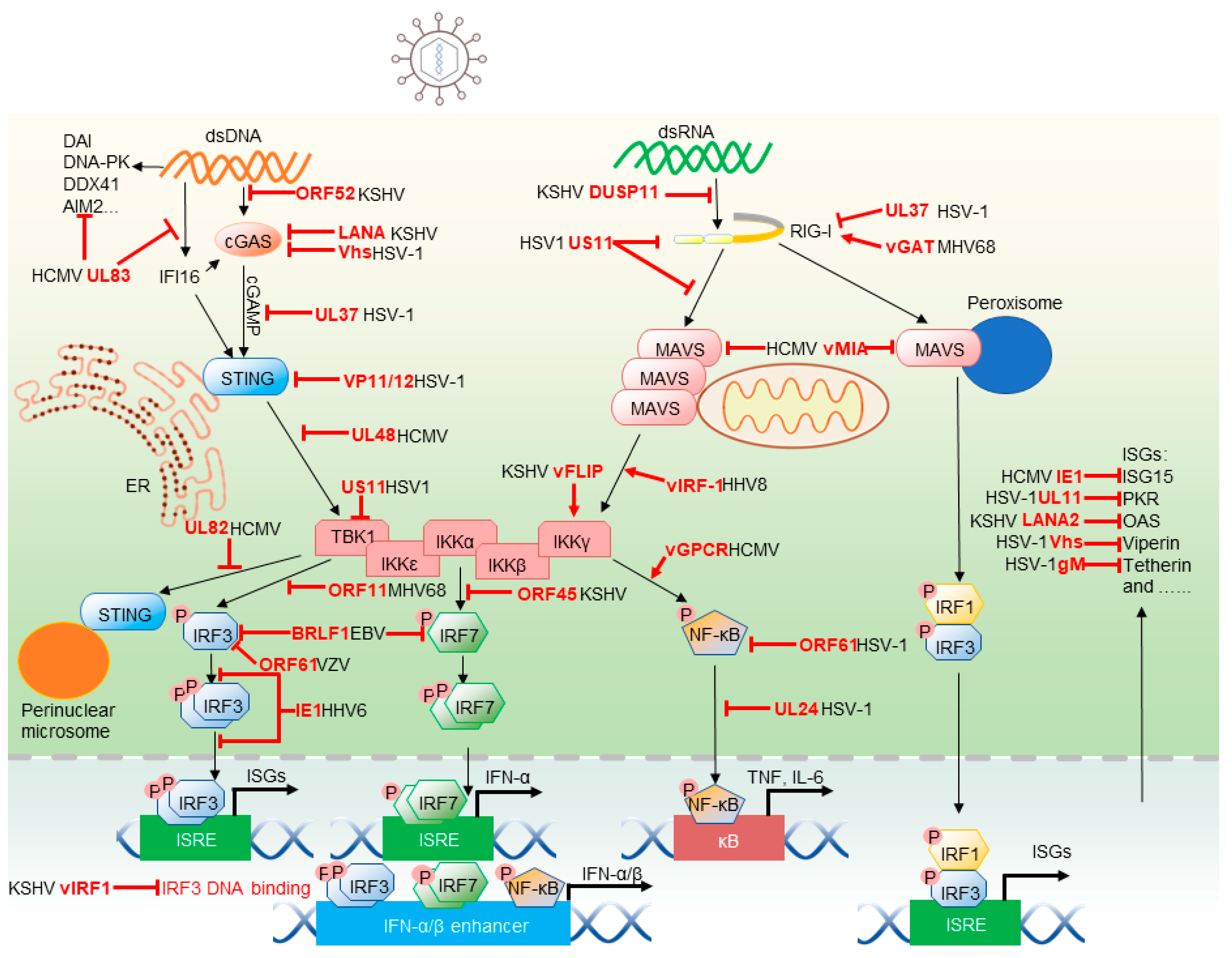
2.1. Cytosolic Sensors and Adaptors
2.1.1. RIG-I-MAVS
2.1.2. cGAS-STING
2.1.3. IFI16
2.1.4. Other Herpesvirus Sensors
2.2. Immune Kinases and Transcscription Factors
2.2.1. IKKβ-NF-κB
2.2.2. TBK1-IRF3
2.3. ISGs with Direct Antiviral Activities
2.3.1. ISG15
2.3.2. Viperin
2.3.3. Tetherin
2.3.4. PKR
2.3.5. OAS
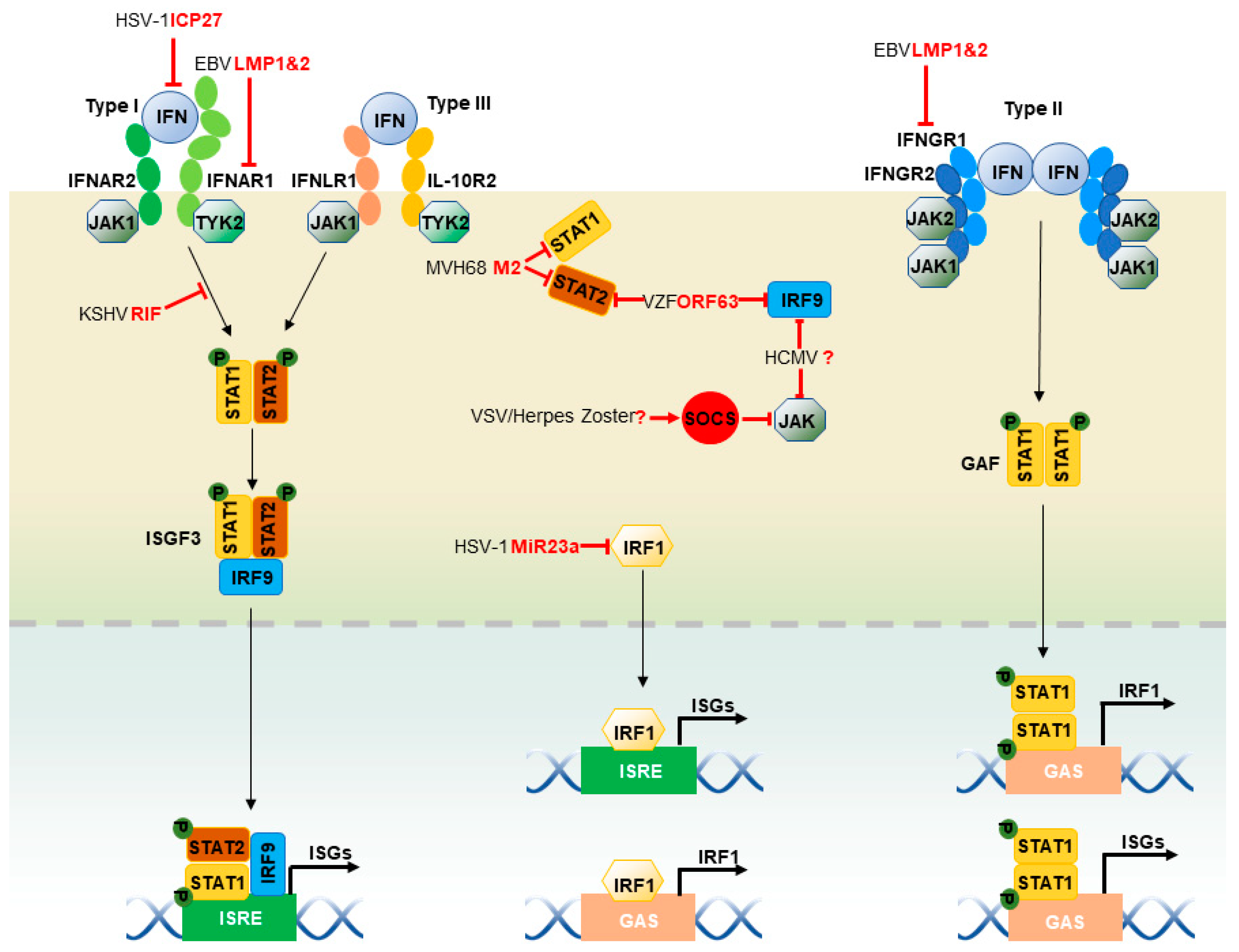
2.4. JAK-STAT Pathway
3. Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields’ Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Ryan, K.J.; Ray, C.G. Medical Microbiology; McGraw Hill: New York, NY, USA, 2004; Volume 4, p. 370. [Google Scholar]
- Roizman, B.; Taddeo, B. The strategy of herpes simplex virus replication and takeover of the host cell. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Melchjorsen, J.; Rintahaka, J.; Søby, S.; Horan, K.A.; Poltajainen, A.; Østergaard, L.; Paludan, S.R.; Matikainen, S. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 2010, 84, 11350–11358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ye, X.; Dunker, W.; Song, Y.; Karijolich, J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun. 2018, 9, 4841. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Bruns, A.M.; Horvath, C.M. LGP2 synergy with MDA5 in RLR-mediated RNA recognition and antiviral signaling. Cytokine 2015, 74, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; e Sousa, C.R. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Wu, B.; Hur, S. How RIG-I like receptors activate MAVS. Curr. Opin. Virol. 2015, 12, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Xu, M.; Liu, S.; Sun, L.; Chen, Z.J. Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol. Cell 2009, 36, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.-T.; Sun, L.; Chen, Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2013, 2, e00785. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of Type I and III Interferon Response by Mitochondrial and Peroxisomal MAVS and Inhibition by Hepatitis C Virus. PLoS Pathog. 2015, 11, e1005264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zeng, Y.; Xu, S.; Chen, J.; Shen, G.; Yu, C.; Knipe, D.; Yuan, W.; Peng, J.; Xu, W. A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 2016, 20, 770–784. [Google Scholar] [CrossRef]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lassig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dittmer, D.P.; Mieczkowski, P.A.; Host, K.M.; Fusco, W.G.; Duncan, J.A.; Damania, B. RIG-I Detects Kaposi’s Sarcoma-Associated Herpesvirus Transcripts in a RNA Polymerase III-Independent Manner. mBio 2018, 9, e00823-18. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, J.; Song, S.; He, X.; Minassian, A.; Zhou, Y.; Zhang, J.; Brulois, K.; Wang, Y.; Cabo, J. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 2015, 58, 134–146. [Google Scholar] [CrossRef]
- Everly, D.N.J.; Feng, P.; Mian, I.S.; Read, G.S. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: Genetic and biochemical evidence that Vhs is a nuclease. J. Virol. 2002, 76, 8560–8571. [Google Scholar] [CrossRef]
- Shiflett, L.A.; Read, G.S. mRNA decay during herpes simplex virus (HSV) infections: Mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein. J. Virol. 2013, 87, 94–109. [Google Scholar] [CrossRef]
- Yao, X.-D.; Rosenthal, K.L. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J. Gen. Virol. 2011, 92, 1981–1993. [Google Scholar] [CrossRef]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef]
- Magalhaes, A.C.; Ferreira, A.R.; Gomes, S.; Vieira, M.; Gouveia, A.; Valenca, I.; Islinger, M.; Nascimento, R.; Schrader, M.; Kagan, J.C.; et al. Peroxisomes are platforms for cytomegalovirus’ evasion from the cellular immune response. Sci. Rep. 2016, 6, 26028. [Google Scholar] [CrossRef]
- Hwang, K.Y.; Choi, Y.B. Modulation of mitochondrial antiviral signaling by human herpesvirus 8 interferon regulatory factor 1. J. Virol. 2016, 90, 506–520. [Google Scholar] [CrossRef]
- Dong, X.; Feng, P. Murine gamma herpesvirus 68 hijacks MAVS and IKKβ to abrogate NFκB activation and antiviral cytokine production. PLoS Pathog. 2011, 7, e1002336. [Google Scholar] [CrossRef]
- Dong, X.; Feng, H.; Sun, Q.; Li, H.; Wu, T.-T.; Sun, R.; Tibbetts, S.A.; Chen, Z.J.; Feng, P. Murine gamma-herpesvirus 68 hijacks MAVS and IKKβ to initiate lytic replication. PLoS Pathog. 2010, 6, e1001001. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef]
- Kranzusch, P.J.; Lee, A.S.; Berger, J.M.; Doudna, J.A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013, 3, 1362–1368. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, X.W.; Jin, J.; Du, X.X.; Lian, T.; Yang, J.; Zhou, X.; Jiang, Z.; Su, X.D. Nonspecific DNA Binding of cGAS N Terminus Promotes cGAS Activation. J. Immunol. 2017, 198, 3627–3636. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Ye, B.; Wang, S.; Zhu, X.; Du, Y.; Xiong, Z.; Tian, Y.; Fan, Z. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 2016, 17, 369–378. [Google Scholar] [CrossRef]
- Hu, M.M.; Yang, Q.; Xie, X.Q.; Liao, C.Y.; Lin, H.; Liu, T.T.; Yin, L.; Shu, H.B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Huang, Y.J.; He, X.; Zhao, M.; Wang, X.; Liu, Z.S.; Xue, W.; Cai, H.; Zhan, X.Y.; Huang, S.Y.; et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell 2019, 176, 1447–1460. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell. Mol. Life Sci. 2011, 68, 1157–1165. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Liu, G.; Lu, Y.; Thulasi Raman, S.N.; Xu, F.; Wu, Q.; Li, Z.; Brownlie, R.; Liu, Q.; Zhou, Y. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 2018, 9, 3199. [Google Scholar] [CrossRef]
- Paijo, J.; Döring, M.; Spanier, J.; Grabski, E.; Nooruzzaman, M.; Schmidt, T.; Witte, G.; Messerle, M.; Hornung, V.; Kaever, V. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog. 2016, 12, e1005546. [Google Scholar] [CrossRef]
- Li, W.; Avey, D.; Fu, B.; Wu, J.-J.; Ma, S.; Liu, X.; Zhu, F. Kaposi’s sarcoma-associated herpesvirus inhibitor of cGAS (KicGAS), encoded by ORF52, is an abundant tegument protein and is required for production of infectious progeny viruses. J. Virol. 2016, 90, 5329–5342. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Kedes, D.H. Mass spectrometric analyses of purified rhesus monkey rhadinovirus reveal 33 virion-associated proteins. J. Virol. 2006, 80, 1574–1583. [Google Scholar] [CrossRef]
- Bortz, E.; Whitelegge, J.P.; Jia, Q.; Zhou, Z.H.; Stewart, J.P.; Wu, T.-T.; Sun, R. Identification of proteins associated with murine gammaherpesvirus 68 virions. J. Virol. 2003, 77, 13425–13432. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.; Robertson, E. KSHV LANA—The master regulator of KSHV latency. Viruses 2014, 6, 4961–4998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chan, B.; Samarina, N.; Abere, B.; Weidner-Glunde, M.; Buch, A.; Pich, A.; Brinkmann, M.M.; Schulz, T.F. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA 2016, 113, E1034–E1043. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-F.; Zou, H.-M.; Liao, B.-W.; Zhang, H.-Y.; Yang, Y.; Fu, Y.-Z.; Wang, S.-Y.; Luo, M.-H.; Wang, Y.-Y. Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to mediate immune evasion. Cell Host Microbe 2018, 24, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M. Human cytomegalovirus tegument protein pp65 (pUL83) dampens type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J. Virol. 2018, 92, e01774-17. [Google Scholar] [CrossRef]
- Jønsson, K.; Laustsen, A.; Krapp, C.; Skipper, K.; Thavachelvam, K.; Hotter, D.; Egedal, J.; Kjolby, M.; Mohammadi, P.; Prabakaran, T. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Almine, J.F.; O’Hare, C.A.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, J.; Xu, S.; Li, J.; He, S.; Zeng, Y.; Xie, L.; Xie, N.; Liu, T.; Lee, K. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 2018, 24, 234–248. [Google Scholar] [CrossRef]
- Su, C.; Zheng, C. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J. Virol. 2017, 91, e02414-16. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, T.; Kalamvoki, M. Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J. Virol. 2017, 91, e00535-17. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Kim, Y.-E.; Stinski, M.F.; Ahn, J.-H.; Song, Y.-J. Human cytomegalovirus IE2 86 kDa protein induces STING degradation and inhibits cGAMP-mediated IFN-β induction. Front. Microbiol. 2017, 8, 1854. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Su, S.; Gao, Y.-Q.; Wang, P.-P.; Huang, Z.-F.; Hu, M.-M.; Luo, W.-W.; Li, S.; Luo, M.-H.; Wang, Y.-Y. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Saha, I.; Narayanan, A.; Narayanan, S.; Takaoka, A.; Kumar, N.S.; Tailor, P.; Kumar, H. Essential role of HCMV deubiquitinase in promoting oncogenesis by targeting anti-viral innate immune signaling pathways. Cell Death Dis. 2017, 8, e3078. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Schattgen, S.A.; Pisitkun, P.; Jorgensen, J.P.; Hilterbrand, A.T.; Wang, L.J.; West, J.A.; Hansen, K.; Horan, K.A.; Jakobsen, M.R. Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J. Immunol. 2015, 194, 1819–1831. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Dell’Oste, V.; Gatti, D.; Gugliesi, F.; De Andrea, M.; Bawadekar, M.; Cigno, I.L.; Biolatti, M.; Vallino, M.; Marschall, M.; Gariglio, M. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 2014, 88, 6970–6982. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [Green Version]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. MBio 2016, 7, e01553-16. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef]
- Cuchet-Lourenço, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear innate immune DNA sensor IFI16 is degraded during lytic reactivation of Kaposi’s sarcoma-associated herpesvirus (KSHV): Role of IFI16 in maintenance of KSHV latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef]
- Schroder, K.; Muruve, D.A.; Tschopp, J. Innate immunity: Cytoplasmic DNA sensing by the AIM2 inflammasome. Curr. Biol. 2009, 19, R262–R265. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Lugrin, J.; Martinon, F. The AIM 2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, D.; Huang, H.; Lu, Y.; Liao, Y.; Liu, L.; Liu, X.; Fang, F. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol. J. 2017, 14, 34. [Google Scholar] [CrossRef]
- Maruzuru, Y.; Ichinohe, T.; Sato, R.; Miyake, K.; Okano, T.; Suzuki, T.; Koshiba, T.; Koyanagi, N.; Tsuda, S.; Watanabe, M. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 2018, 23, 254–265. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.-W.; Lempicki, R.A.; Baseler, M.W. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Tognarelli, E.I.; Palomino, T.F.; Corrales, N.; Bueno, S.M.; Kalergis, A.M.; Gonzalez, P.A. Herpes Simplex Virus Evasion of Early Host Antiviral Responses. Front. Cell. Infect. Microbiol. 2019, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Carriere, J.; Lin, X.; Xie, N.; Feng, P. Interplay between cellular metabolism and cytokine responses during viral infection. Viruses 2018, 10, 521. [Google Scholar] [CrossRef]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Guo, B.; Cheng, G. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J. Biol. Chem. 2007, 282, 11817–11826. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Mukherjee, S.P.; Behar, M.; Birnbaum, H.A.; Hoffmann, A.; Wright, P.E.; Ghosh, G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 2013, 11, e1001647. [Google Scholar] [CrossRef] [PubMed]
- Wienerroither, S.; Shukla, P.; Farlik, M.; Majoros, A.; Stych, B.; Vogl, C.; Cheon, H.; Stark, G.R.; Strobl, B.; Muller, M.; et al. Cooperative Transcriptional Activation of Antimicrobial Genes by STAT and NF-kappaB Pathways by Concerted Recruitment of the Mediator Complex. Cell Rep. 2015, 12, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Guasparri, I.; Keller, S.A.; Cesarman, E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 2004, 199, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Eby, M.T.; Rathore, N.; Sinha, S.K.; Kumar, A.; Chaudhary, P.M. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the IκB kinase complex. J. Biol. Chem. 2002, 277, 13745–13751. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Podgrabinska, S.; Skobe, M.; Ganem, D. Activation of NF-κB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J. Virol. 2006, 80, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Ersing, I.; Bernhardt, K.; Gewurz, B. NF-κB and IRF7 pathway activation by Epstein-Barr virus latent membrane protein 1. Viruses 2013, 5, 1587–1606. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, B.C.; Blonska, M.; Lin, P.-C.; You, Y.; Wang, D.; Sun, J.; Darnay, B.G.; Dong, C.; Lin, X. CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-κB activation. Genes Dev. 2007, 21, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Blonska, M.; Lin, X. NF-κB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 2011, 21, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chung, Y.H.; Lee, C. US28, a virally-encoded GPCR as an antiviral target for human cytomegalovirus infection. Biomol. Ther. 2017, 25, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Philpott, N.; Bakken, T.; Pennell, C.; Chen, L.; Wu, J.; Cannon, M. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor contains an immunoreceptor tyrosine-based inhibitory motif that activates Shp2. J. Virol. 2011, 85, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 abrogates NF-κB activation in DNA sensing signal pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, T.; Malouli, D.; Uebelhoer, L.S.; DeFilippis, V.R.; Früh, K.; Verweij, M.C. The ORF61 protein encoded by simian varicella virus and varicella-zoster virus inhibits NF-κB signaling by interfering with IκBα degradation. J. Virol. 2015, 89, 8687–8700. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Su, C.; Pearson, A.; Mody, C.H.; Zheng, C. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-κB activation. J. Virol. 2017, 91, e00025-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Wang, K.; Zheng, C. Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-α-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1. Med Microbiol. Immunol. 2013, 202, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.; Schafer, X.; Martínez-Sobrido, L.; Munger, J. The human cytomegalovirus UL26 protein antagonizes NF-κB activation. J. Virol. 2014, 88, 14289–14300. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef]
- Kang, H.-R.; Cheong, W.-C.; Park, J.-E.; Ryu, S.; Cho, H.-J.; Youn, H.; Ahn, J.-H.; Song, M.J. Murine gammaherpesvirus 68 encoding open reading frame 11 targets TANK binding kinase 1 to negatively regulate the host type I interferon response. J. Virol. 2014, 88, 6832–6846. [Google Scholar] [CrossRef]
- Liu, X.; Main, D.; Ma, Y.; He, B. Herpes simplex virus 1 inhibits TANK-binding kinase 1 through formation of the Us11-Hsp90 complex. J. Virol. 2018, 92, e00402-18. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C.; Xing, J.; Wang, S.; Li, S.; Lin, R.; Mossman, K.L. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J. Virol. 2011, 85, 11079–11089. [Google Scholar] [CrossRef]
- Sen, N.; Sommer, M.; Che, X.; White, K.; Ruyechan, W.T.; Arvin, A.M. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: A novel mechanism of IRF3 inhibition among herpesviruses. J. Virol. 2010, 84, 9240–9253. [Google Scholar] [CrossRef]
- Vandevenne, P.; Lebrun, M.; El Mjiyad, N.; Ote, I.; Di Valentin, E.; Habraken, Y.; Dortu, E.; Piette, J.; Sadzot-Delvaux, C. The varicella-zoster virus ORF47 kinase interferes with host innate immune response by inhibiting the activation of IRF3. PLoS ONE 2011, 6, e16870. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Gravel, A.; Fink, K.; Grandvaux, N.; Flamand, L. Inhibition of transcription of the beta interferon gene by the human herpesvirus 6 immediate-early 1 protein. J. Virol. 2007, 81, 5737–5748. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A.; Harrington, W.J.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.; Kim, K.S.; Flano, E.; Wu, T.-T.; Tong, L.M.; Park, A.N.; Song, M.J.; Sanchez, D.J.; O’Connell, R.M.; Cheng, G. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 2009, 5, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Soucy-Faulkner, A.; Grandvaux, N.; Flamand, L. Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 2007, 81, 10950–10960. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-T.; Doong, S.-L.; Teng, S.-C.; Lee, C.-P.; Tsai, C.-H.; Chen, M.-R. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 2009, 83, 1856–1869. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; King, S.M.; Smith, E.J.; Levy, D.E.; Yuan, Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. USA 2002, 99, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.M.; Huye, L.E.; Ning, S.; Webster-Cyriaque, J.; Pagano, J.S. Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene, BZLF-1. J. Virol. 2005, 79, 10040–10052. [Google Scholar] [CrossRef] [PubMed]
- Bentz, G.L.; Liu, R.; Hahn, A.M.; Shackelford, J.; Pagano, J.S. Epstein–Barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-β. Virology 2010, 402, 121–128. [Google Scholar] [CrossRef]
- Malakhov, M.P.; Malakhova, O.A.; Kim, K.I.; Ritchie, K.J.; Zhang, D.-E. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J. Biol. Chem. 2002, 277, 9976–9981. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Guerra, S.; Sánchez-Madrid, F. ISGylation–a key to lock the cell gates for preventing the spread of threats. J. Cell Sci. 2017, 130, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.-X.; Yang, K.; Liu, X.; Liu, X.-Y.; Wei, B.; Shan, Y.-F.; Zhu, L.-H.; Wang, C. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell. Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, E.T.; Kim, Y.E.; Lee, M.K.; Kwon, K.M.; Kim, K.I.; Stamminger, T.; Ahn, J.H. Consecutive Inhibition of ISG15 Expression and ISGylation by Cytomegalovirus Regulators. PLoS Pathog. 2016, 12, e1005850. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Stopford, C.M.; West, J.A.; Bennett, C.L.; Giffin, L.; Damania, B. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 1 Interacts with a Member of the Interferon-Stimulated Gene 15 Pathway. J. Virol. 2015, 89, 11572–11583. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Beard, M.R. The role of viperin in the innate antiviral response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef]
- Ng, L.F.; Hiscox, J.A. Viperin Poisons Viral Replication. Cell Host Microbe 2018, 24, 181–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gizzi, A.S.; Grove, T.L.; Arnold, J.J.; Jose, J.; Jangra, R.K.; Garforth, S.J.; Du, Q.; Cahill, S.M.; Dulyaninova, N.G.; Love, J.D. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 2018, 558, 610–614. [Google Scholar] [CrossRef]
- Dumbrepatil, A.B.; Ghosh, S.; Zegalia, K.A.; Malec, P.A.; Hoff, J.D.; Kennedy, R.T.; Marsh, E.N.G. Viperin interacts with the kinase IRAK1 and the E3 ubiquitin ligase TRAF6, coupling innate immune signaling to antiviral ribonucleotide synthesis. J. Biol. Chem. 2019, 294, 6888–6898. [Google Scholar] [CrossRef]
- Shen, G.; Wang, K.; Wang, S.; Cai, M.; Li, M.-L.; Zheng, C. Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein UL41. J. Virol. 2014, 88, 12163–12166. [Google Scholar] [CrossRef]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef]
- Swiecki, M.; Omattage, N.S.; Brett, T.J. BST-2/tetherin: Structural biology, viral antagonism, and immunobiology of a potent host antiviral factor. Mol. Immunol. 2013, 54, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.T.; Serra-Moreno, R.; Singh, R.K.; Guatelli, J.C. BST-2/tetherin: A new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010, 18, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, C.; Pelchen-Matthews, A.; Mlcochova, P.; Marsh, M.; Milne, R.S.; Towers, G.J. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J. Virol. 2013, 87, 13124–13133. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Viswanathan, K.; Douglas, J.L.; Hines, J.; Gustin, J.; Moses, A.V.; Fruh, K. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2009, 83, 9672–9681. [Google Scholar] [CrossRef] [PubMed]
- Green, S.R.; Mathews, M.B. Two RNA-binding motifs in the double-stranded RNA-activated protein kinase, DAI. Genes Dev. 1992, 6, 2478–2490. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Sen, G.C. Identification of the double-stranded RNA-binding domain of the human interferon-inducible protein kinase. J. Biol. Chem. 1992, 267, 7671–7676. [Google Scholar] [PubMed]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.B.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Galabru, J.; Hovanessian, A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J. Biol. Chem. 1987, 262, 15538–15544. [Google Scholar] [PubMed]
- Samuel, C.E. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 1993, 268, 7603–7606. [Google Scholar]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-κB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-κB-inducing kinase and IκB kinase. Mol. Cell. Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Khoo, D.; Mohr, I.; Sen, G.C. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 2002, 76, 11054–11064. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Corrigendum: Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2015, 525, 144. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.; Garcia, M.; Domingo-Gil, E.; Arroyo, J.; Nombela, C.; Rivas, C. The latency protein LANA2 from Kaposi’s sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol. 2003, 84, 1463–1470. [Google Scholar] [CrossRef]
- Pulit-Penaloza, J.A.; Scherbik, S.V.; Brinton, M.A. Type 1 IFN-independent activation of a subset of interferon stimulated genes in West Nile virus Eg101-infected mouse cells. Virology 2012, 425, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-J.; Latham, A.G.; Krug, R.M. Human influenza viruses activate an interferon-independent transcription of cellular antiviral genes: Outcome with influenza A virus is unique. Proc. Natl. Acad. Sci. USA 2002, 99, 10096–10101. [Google Scholar] [CrossRef] [Green Version]
- Hovanessian, A.G. Interferon-induced and double-stranded RNA-activated enzymes: A specific protein kinase and 2′, 5′-oligoadenylate synthetases. J. Interferon Res. 1991, 11, 199–205. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Jha, B.K.; Silverman, R.H. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 2011, 31, 49–57. [Google Scholar] [CrossRef]
- Silverman, R.H. Viral encounters with 2′, 5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef]
- Rebouillat, D.; Hovanessian, A.G. The human 2′, 5′-oligoadenylate synthetase family: Interferon-induced proteins with unique enzymatic properties. J. Interferon Cytokine Res. 1999, 19, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale Jr, M.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Shao, L.; Sampath, P.; Zhao, B.; Patel, N.V.; Zhu, J.; Behl, B.; Parise, R.A.; Beumer, J.H.; O’Sullivan, R.J.; et al. Oligoadenylate-Synthetase-Family Protein OASL Inhibits Activity of the DNA Sensor cGAS during DNA Virus Infection to Limit Interferon Production. Immunity 2019, 50, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Carey, C.M.; Govande, A.A.; Cooper, J.M.; Hartley, M.K.; Kranzusch, P.J.; Elde, N.C. Recurrent loss-of-function mutations reveal costs to OAS1 antiviral activity in primates. Cell Host Microbe 2019, 25, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Sànchez, R.; Mohr, I. Inhibition of cellular 2′-5′ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 2007, 81, 3455–3464. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wu, C.; Zhang, Y.-J. Interplay between Janus kinase/signal transducer and activator of transcription signaling activated by type I interferons and viral antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: Development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines 2016, 4, 23. [Google Scholar] [CrossRef]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.; O’neil, J.; Dawson, C.W.; Young, L.S. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Shin, Y.C.; Means, R.E.; Jung, J.U. Inhibition of interferon-mediated antiviral activity by murine gammaherpesvirus 68 latency-associated M2 protein. J. Virol. 2004, 78, 12416–12427. [Google Scholar] [CrossRef]
- Morrison, T.E.; Mauser, A.; Wong, A.; Ting, J.P.-Y.; Kenney, S.C. Inhibition of IFN-γ signaling by an Epstein-Barr virus immediate-early protein. Immunity 2001, 15, 787–799. [Google Scholar] [CrossRef]
- Johnson, K.E.; Knipe, D.M. Herpes simplex virus-1 infection causes the secretion of a type I interferon-antagonizing protein and inhibits signaling at or before Jak-1 activation. Virology 2010, 396, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisson, S.A.; Page, A.-L.; Ganem, D. A Kaposi’s sarcoma-associated herpesvirus protein that forms inhibitory complexes with type I interferon receptor subunits, Jak and STAT proteins, and blocks interferon-mediated signal transduction. J. Virol. 2009, 83, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Lee, C.H.; Shin, O. Suppressor of Cytokine Signaling 3 Expression Induced by Varicella-Zoster Virus Infection Results in the Modulation of Virus Replication. Scand. J. Immunol. 2015, 82, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Zhang, Y.; Rahill, B.M.; Waldman, W.J.; Sedmak, D.D. Human cytomegalovirus inhibits IFN-α-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-α signal transduction. J. Immunol. 1999, 162, 6107–6113. [Google Scholar] [PubMed]
- Verweij, M.C.; Wellish, M.; Whitmer, T.; Malouli, D.; Lapel, M.; Jonjić, S.; Haas, J.G.; DeFilippis, V.R.; Mahalingam, R.; Früh, K. Varicella viruses inhibit interferon-stimulated JAK-STAT signaling through multiple mechanisms. PLoS Pathog. 2015, 11, e1004901. [Google Scholar] [CrossRef] [PubMed]
- Kröger, A.; Köster, M.; Schroeder, K.; Hauser, H.; Mueller, P.P. Activities of IRF-1. J. Interferon Cytokine Res. 2002, 22, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Pine, R.; Decker, T.; Kessler, D.; Levy, D.; Darnell, J. Purification and cloning of interferon-stimulated gene factor 2 (ISGF2): ISGF2 (IRF-1) can bind to the promoters of both beta interferon-and interferon-stimulated genes but is not a primary transcriptional activator of either. Mol. Cell. Biol. 1990, 10, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Kovarik, P.; Meinke, A. GAS elements: A few nucleotides with a major impact on cytokine-induced gene expression. J. Interferon Cytokine Res. 1997, 17, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; Kirchhoff, S.; Posern, G.; Köster, M.; Oumard, A.; Sharf, R.; Ben-Zion, L.; Hauser, H. Functional domains of interferon regulatory factor I (IRF-1). Biochem. J. 1998, 335, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee-Kishore, M.; Wright, K.L.; Ting, J.P.Y.; Stark, G.R. How Stat1 mediates constitutive gene expression: A complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000, 19, 4111–4122. [Google Scholar] [CrossRef]
- El Hassan, M.A.; Huang, K.; Eswara, M.B.; Xu, Z.; Yu, T.; Aubry, A.; Ni, Z.; Livne-Bar, I.; Sangwan, M.; Ahmad, M. Properties of STAT1 and IRF1 enhancers and the influence of SNPs. BMC Mol. Biol. 2017, 18, 6. [Google Scholar] [CrossRef]
- Ru, J.; Sun, H.; Fan, H.; Wang, C.; Li, Y.; Liu, M.; Tang, H. MiR-23a facilitates the replication of HSV-1 through the suppression of interferon regulatory factor 1. PLoS ONE 2014, 9, e114021. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Rao, Y.; Tian, M.; Zhang, S.; Feng, P. Modulation of Innate Immune Signaling Pathways by Herpesviruses. Viruses 2019, 11, 572. https://doi.org/10.3390/v11060572
Liu Q, Rao Y, Tian M, Zhang S, Feng P. Modulation of Innate Immune Signaling Pathways by Herpesviruses. Viruses. 2019; 11(6):572. https://doi.org/10.3390/v11060572
Chicago/Turabian StyleLiu, Qizhi, Youliang Rao, Mao Tian, Shu Zhang, and Pinghui Feng. 2019. "Modulation of Innate Immune Signaling Pathways by Herpesviruses" Viruses 11, no. 6: 572. https://doi.org/10.3390/v11060572
APA StyleLiu, Q., Rao, Y., Tian, M., Zhang, S., & Feng, P. (2019). Modulation of Innate Immune Signaling Pathways by Herpesviruses. Viruses, 11(6), 572. https://doi.org/10.3390/v11060572