Evidence of Intragenic Recombination in African Horse Sickness Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sets
2.2. Generating Complete Genome Sequences of African Horse Sickness Viruses
2.3. Phylogenetic and Recombination Analysis
3. Results
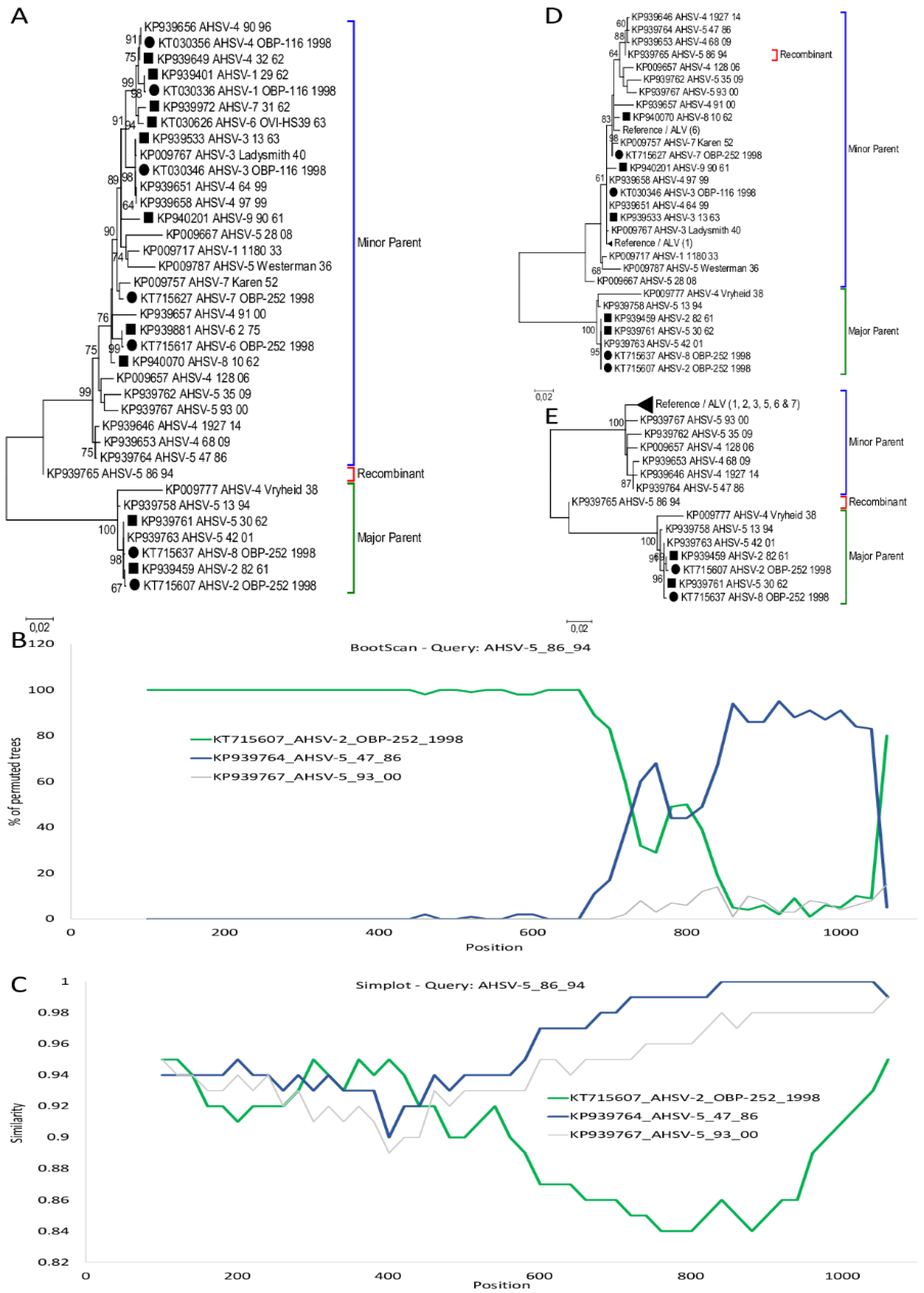
3.1. Segment-1 (VP1)
3.2. Segment-6 (VP5)
3.3. Segment-7 (VP7)
3.4. Segment-10 (NS3)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Theiler, A. African horse sickness (Pestis Equorum). Sci. Bull. 1921, 19, 1–29. [Google Scholar]
- Mellor, P.S.; Hamblin, C. African horse sickness. Vet. Res. 2004, 35, 445–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Toit, R.M. The transmission of bluetongue and horse-sickness by Culicoides. Onderstepoort J. Vet. Sci. Anim. Ind. 1944, 10, 7–17. [Google Scholar]
- Meiswinkel, R.; Paweska, J.T. Evidence for a new field Culicoides vector of African horsesickness in South Africa. Prev. Vet. Med. 2003, 60, 243–253. [Google Scholar] [CrossRef]
- Sinclair, M.; Buhrmann, G.; Gummow, B. An epidemiological investigation of the African horsesickness outbreak in the Western Cape Province of South Africa in 2004 and its relevance to the current equine export protocol. J. S. Afr. Vet. Assoc. 2006, 77, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, A.; Deeks, D. Electron microscopy of neurotopic African horse-sickness virus. J. Hyg. 1963, 61, 149153. [Google Scholar] [CrossRef]
- Bremer, C.W. A gel electrophoretic study of the protein and nucleic acid components of African horsesickness virus. Onderstepoort J. Vet. Res. 1976, 43, 193–200. [Google Scholar]
- Bremer, C.W.; Huisman, H.; Van Dijk, A.A. Characterization and cloning of the African horsesickness virus genome. J. Gen. Virol. 1990, 71, 793–799. [Google Scholar] [CrossRef]
- Grubman, M.J.; Lewis, S.A. Identification and characterization of the structural and non-structural proteins of African horsesickness virus and determination of the genome coding assignments. Virology 1992, 186, 444–451. [Google Scholar] [CrossRef]
- Ratinier, M.; Caporale, M.; Golder, M.; Franzoni, G.; Allan, K.; Nunes, S.F.; Armezzani, A.; Bayoumy, A.; Rixon, F.; Shaw, A.; et al. Identification and characterization of a novel non-structural protein of bluetongue virus. PLoS Pathog. 2011, 7, e1002477. [Google Scholar] [CrossRef]
- Zwart, L.; Potgieter, C.A.; Clift, S.J.; van Staden, V. Characterizing non-structural protein NS4 of African horse sickness virus. PLoS ONE 2015, 10, e0124281. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, B.M. Immunological types of horsesickness virus and their significance in immunization. Onderstepoort J. Vet. Res. 1958, 27, 465–538. [Google Scholar]
- Howell, P.G. The isolation and identification of further antigenic types of African horsesickness virus. Onderstepoort J. Vet. Res. 1963, 29, 139149. [Google Scholar]
- Martınez-Torrecuadrada, J.L.; Iwata, H.; Venteo, A.; Casal, I.; Roy, P. Expression and characterization of the two outer capsid proteins of African horsesickness virus: The role of VP2 in virus neutralization. Virology 1994, 202, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Mertens, P.P.; Casal, I. African horsesickness virus structure. Comp. Immunol. Microbiol. Infect. Dis. 1994, 17, 243–273. [Google Scholar] [CrossRef]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- He, C.Q.; Ding, N.Z.; He, M.; Li, S.N.; Wang, X.M.; He, H.B.; Liu, X.F.; Guo, H.S. Intragenic recombination as a mechanism of genetic diversity in bluetongue virus. J. Virol. 2010, 84, 11487–11495. [Google Scholar] [CrossRef]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis revelas a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Mechanisms of viral emergence. Vet. Res. 2010, 41, 38. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.D.; Simon, A.E. New insights into the mechanisms of RNA recombination. Virology 1997, 235, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R.; Roze, D.; Uzest, M.; Galibert, L.; Blanc, S.; Michalakis, Y. Recombination every day: Abundant recombination in a virus during a single multi-cellular host infection. PLoS Biol. 2005, 3, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Perez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.J. Intrasegmental recombination does not contribute to the long-term evolution of group A rotavirus. Infect. Genet. Evol. 2015, 32, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.T.; Grewar, J.D.; Burger, P.; Rossouw, E.; Lourens, C.; Joone, C.; le Grange, M.; Coetzee, P.; Venter, E.; Martin, D.P.; et al. African horse sickness caused by genome reassortment and reversion to virulence of live, attenuated vaccine viruses, South Africa, 2004–2014. Emerg. Infect. Dis. 2016, 22, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Potgieter, A.C.; Wright, I.M.; van Dijk, A.A. Consensus Sequence of 27 African Horse Sickness Virus Genomes from Viruses Collected over a 76-Year Period (1933 to 2009). Genome Announc. 2015, 3, e00921-15. [Google Scholar] [CrossRef] [Green Version]
- Guthrie, A.J.; Coetzee, P.; Martin, D.P.; Lourens, C.W.; Venter, E.H.; Weyer, C.T.; Joone, C.; le Grange, M.; Harper, C.K.; Howell, P.G.; et al. Complete genome sequences of the three African horse sickness virus strains from a commercial trivalent live attenuated vaccine. Genome Announc. 2015, 3, e00814-15. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, A.J.; Coetzee, P.; Martin, D.P.; Lourens, C.W.; Venter, E.H.; Weyer, C.T.; Joone, C.; le Grange, M.; Harper, C.K.; Howell, P.G.; et al. Complete genome sequences of the four African horse sickness virus strains from a commercial tetravalent live attenuated vaccine. Genome Announc. 2015, 3, e01375-15. [Google Scholar] [CrossRef]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; van Dijk, A.A. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 2009, 90, 1423–1432. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Heath, L.; van der Walt, E.; Varsani, A.; Martin, D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified BOOTSCAN algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawye, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected sero-converters in India, with evidence of inter-subtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- Quan, M.; van Vuuren, M.; Howell, P.G.; Groenewald, D.; Guthrie, A.J. Molecular epidemiology of the African horse sickness virus S10 gene. J. Gen. Virol. 2008, 89, 1159–1168. [Google Scholar] [CrossRef]
- Lai, M.M. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar]
- Walker, P.J.; Cowley, J.A. Viral Genetic Variation: Implications for Disease Diagnosis and Detection of Shrimp Pathogens; FAO: Rome, Italy, 2000; Volume 395, pp. 54–59. [Google Scholar]
- Urakawa, T.; Ritter, D.G.; and Roy, P. Expression of largest RNA segment and synthesis of VP1 protein of bluetongue virus in insect cells by recombinant baculovirus: Association of VP1 protein with RNA polymerase activity. Nucleic Acids Res. 1989, 17, 7395–7401. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, B.J. A new approach to polyvalent immunization against African horse sickness. J. Equine Med. Surg. 1978, 1, 401–403. [Google Scholar]
- Weyer, C.T.; Joone, C.; Lourens, C.W.; Monyai, M.S.; Koekemoer, O.; Grewar, J.D.; van Schalkwyk, A.; Majiwa, P.O.; MacLachlan, N.J.; Guthrie, A.J. Development of three triplex real-time reverse transcription PCR assays for the qualitative molecular typing of the nine serotypes of African horse sickness virus. J. Virol. Methods 2015, 223, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, S.H.; Wirblich, C.; Forzan, M.; Roy, P. Expression and functional characterization of bluetongue virus VP5 protein: Role in cellular permeabilization. J. Virol. 2001, 75, 8356–8367. [Google Scholar] [CrossRef] [PubMed]
- Basak, A.K.; Gouet, P.; Roy, P.; Stuart, D. Crystal structure of the top domain of African horse sickness virus VP7, comparisons with Bluetongue virus VP7. J. Virol. 1996, 70, 3797–3806. [Google Scholar] [PubMed]
- Sailleau, C.; Moulay, S.; Zientara, S. Nucleotide sequence comparison of the segments S10 of the nine African horse sickness virus serotypes. Arch. Virol. 1997, 142, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.A.; Meyer, A.J.; O’Hara, R.S.; Fu, H.; Mellor, P.S.; Knowles, N.J.; Mertens, P.P. Phylogenetic analysis of African horse sickness virus segment 10: Sequence variation, virulence characteristics and cell exit. Arch. Virol. 1998, 14, 281–293. [Google Scholar]
- Van Niekerk, M.; Smit, C.C.; Fick, W.C.; van Staden, V.; Huismans, H. Membrane association of African horsesickness virus nonstructural protein NS3 determines its cytotoxicity. Virology 2001, 279, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Jenckel, M.; Bréard, E.; Schulz, C.; Sailleau, C.; Viarouge, C.; Hoffmann, B.; Höper, D.; Beer, M.; Zientara, S. Complete coding genome sequence of putative novel bluetongue virus serotype 27. Genome Announc. 2015, 12, e00016-15. [Google Scholar] [CrossRef]
- Maclachlan, N.J.; Guthrie, A.J. Re-emergence of bluetongue, African horse sickness and other orbivirus diseases. Vet. Res. 2010, 41, 35. [Google Scholar] [CrossRef]
- Von Teichman, B.F.; Dungu, B.; Smith, T.K. In vivo cross-protection of African horse sickness serotype 5 and 9 after vaccination with serotypes 8 and 6. Vaccine 2010, 28, 6505–6517. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngoveni, H.G.; van Schalkwyk, A.; Koekemoer, J.J.O. Evidence of Intragenic Recombination in African Horse Sickness Virus. Viruses 2019, 11, 654. https://doi.org/10.3390/v11070654
Ngoveni HG, van Schalkwyk A, Koekemoer JJO. Evidence of Intragenic Recombination in African Horse Sickness Virus. Viruses. 2019; 11(7):654. https://doi.org/10.3390/v11070654
Chicago/Turabian StyleNgoveni, Harry G., Antoinette van Schalkwyk, and J.J. Otto Koekemoer. 2019. "Evidence of Intragenic Recombination in African Horse Sickness Virus" Viruses 11, no. 7: 654. https://doi.org/10.3390/v11070654
APA StyleNgoveni, H. G., van Schalkwyk, A., & Koekemoer, J. J. O. (2019). Evidence of Intragenic Recombination in African Horse Sickness Virus. Viruses, 11(7), 654. https://doi.org/10.3390/v11070654