Genetic Variability of Chikungunya Virus in Southern Mexico

 , ,
, ,
Abstract
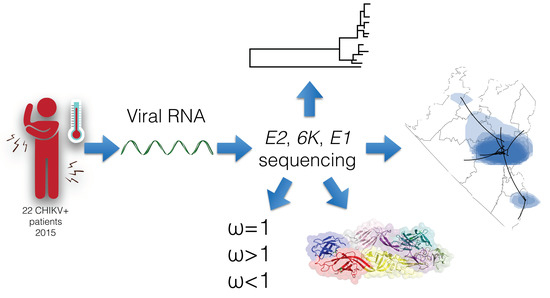
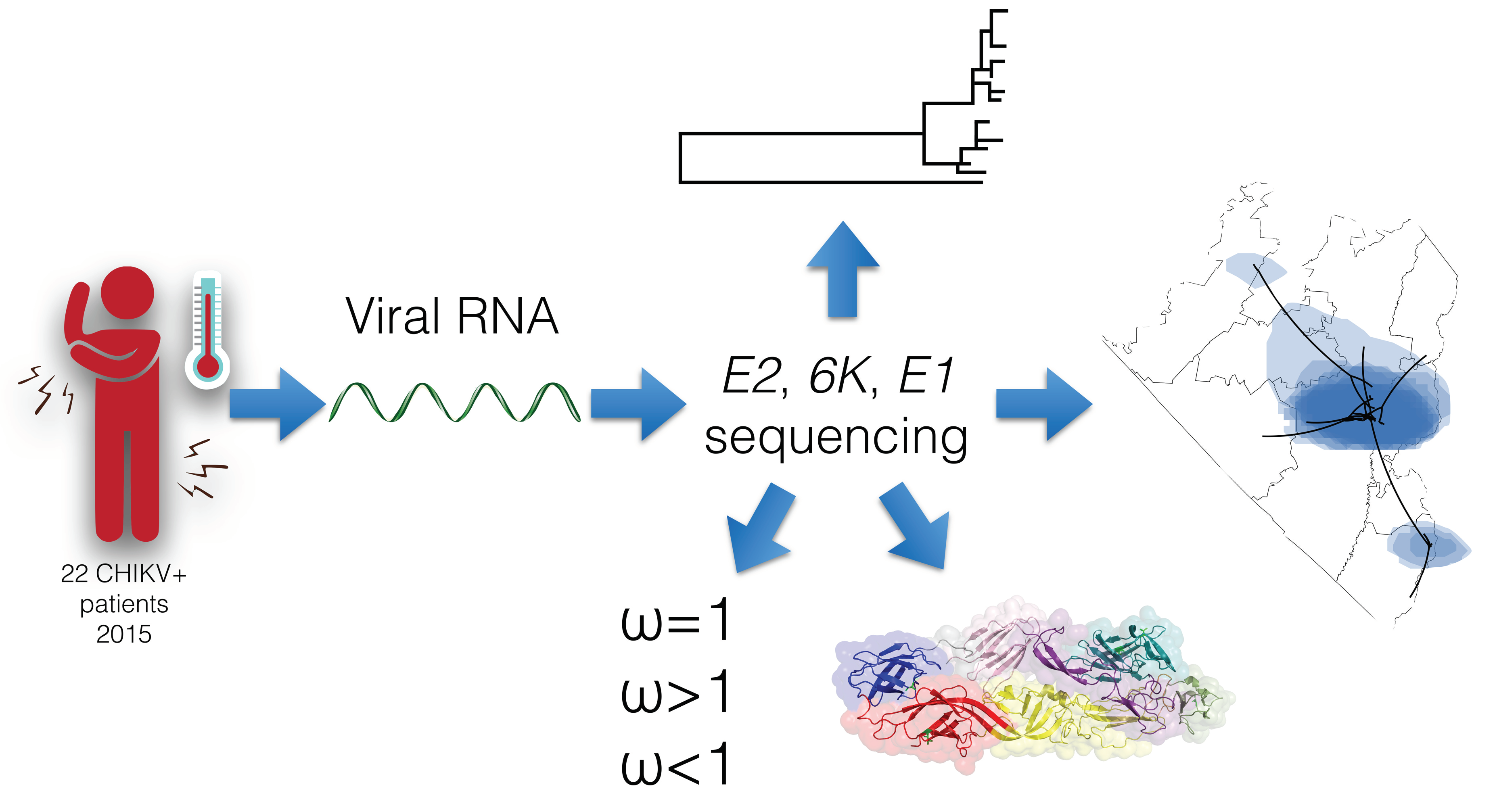
:
1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Study Population
2.3. Viral RNA Detection
2.4. Envelope Genes Amplification and Sequencing
2.5. Data Sets
2.6. Phylogenetic Analysis
2.7. Phylogeographic Inference
2.8. Molecular Modeling
2.9. Selection Analyses
3. Results
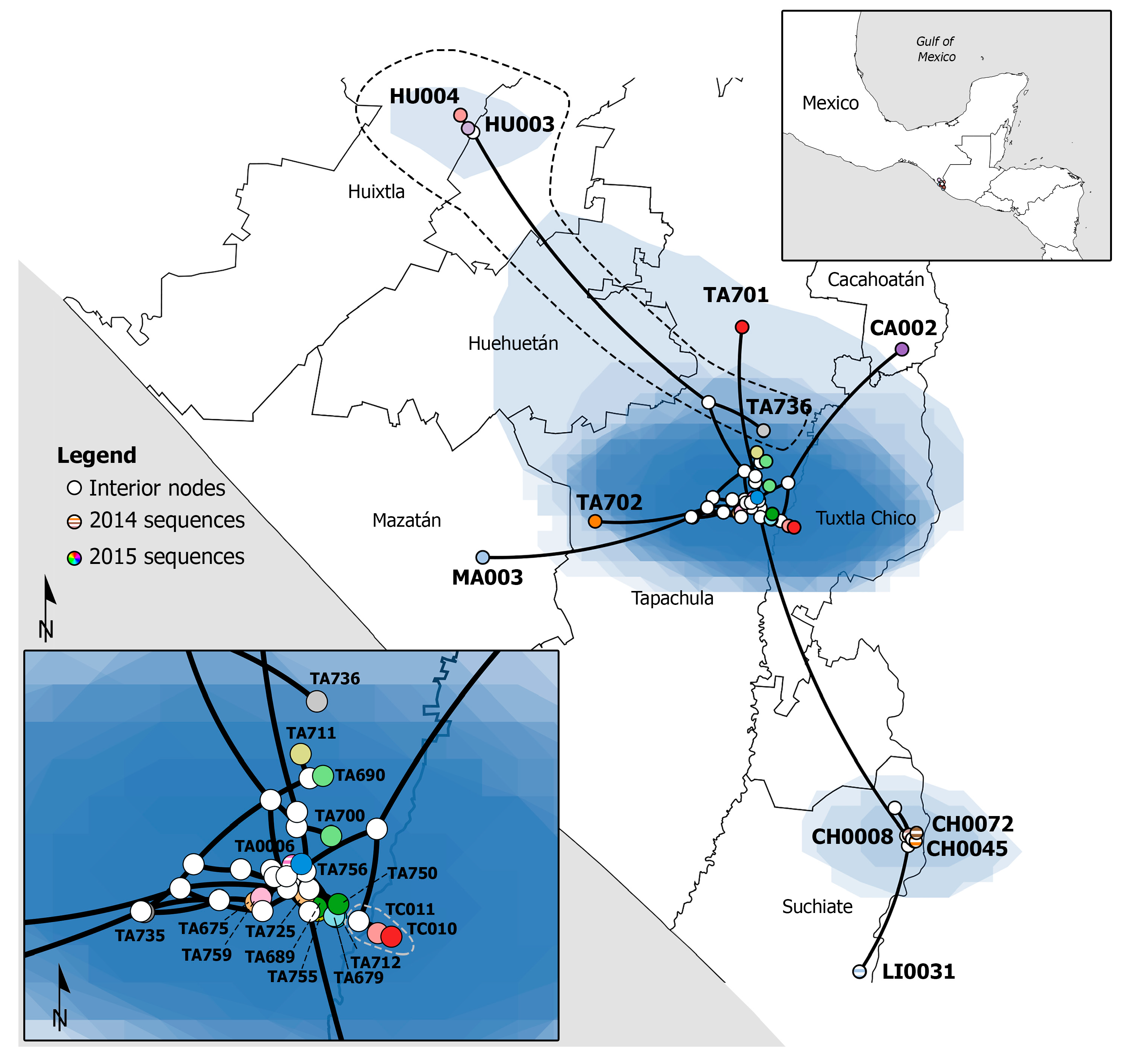
3.1. Phylogenetic Analysis and Phylogeographic Inference
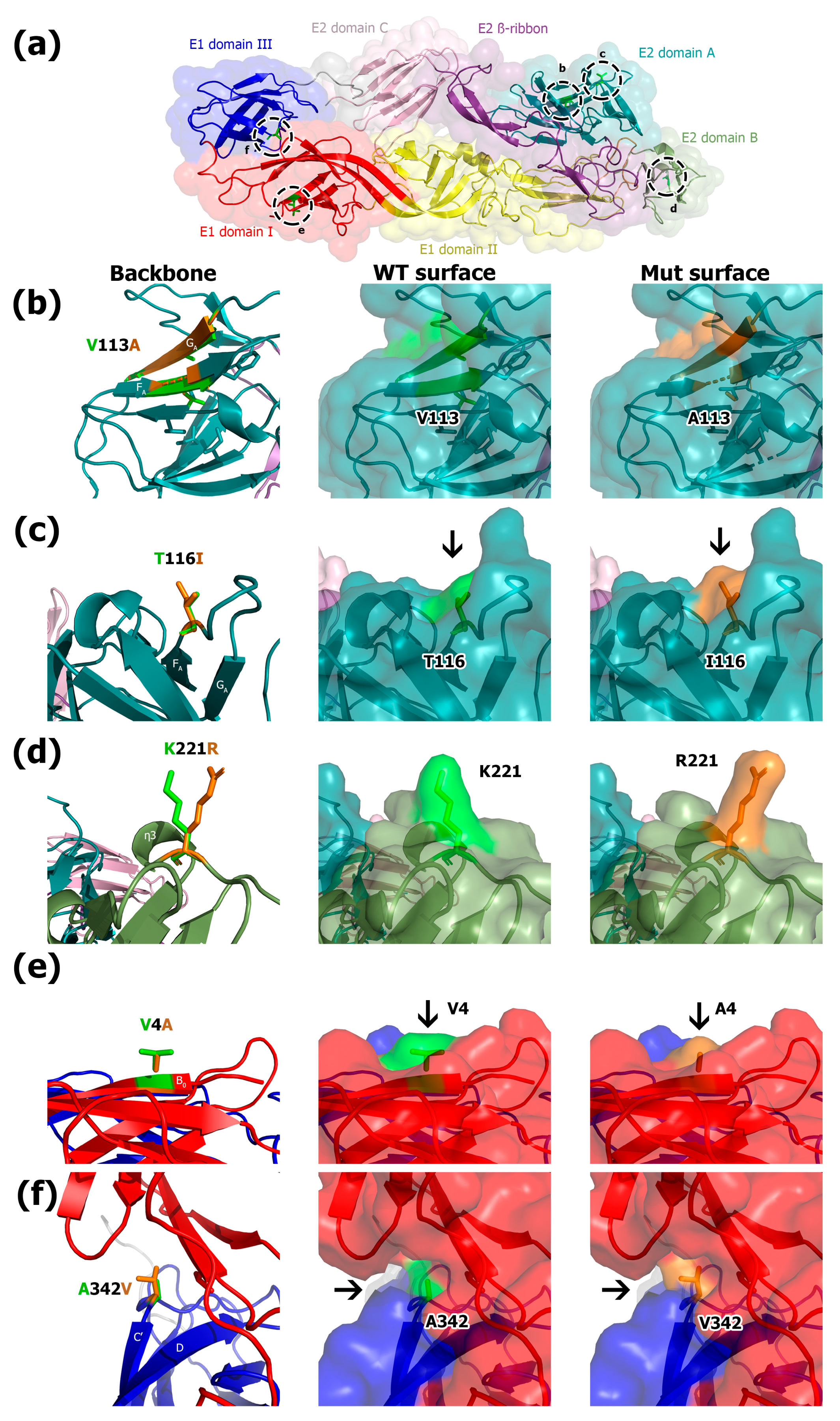
3.2. Molecular Modeling
3.3. Selection Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weaver, S.C.; Lecuit, M. Chikungunya Virus and the Global Spread of a Mosquito-Borne Disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Caglioti, C.; Lalle, E.; Castilletti, C.; Carletti, F.; Capobianchi, M.R.; Bordi, L. Chikungunya virus infection: An overview. New Microbiol. 2013, 36, 211–227. [Google Scholar] [PubMed]
- Schwartz, O.; Albert, M.L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Burt, F.J.; Chen, W.; Miner, J.J.; Lenschow, D.J.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya virus: An update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Griffin, D. Alphaviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013; p. 655. ISBN 1-4511-0563-0. [Google Scholar]
- Voss, J.E.; Vaney, M.-C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef]
- Zhang, R.; Hryc, C.F.; Cong, Y.; Liu, X.; Jakana, J.; Gorchakov, R.; Baker, M.L.; Weaver, S.C.; Chiu, W. 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 2011, 30, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Yun, R.; Rossi, S.L.; Plante, K.S.; Guerbois, M.; Forrester, N.; Perng, G.C.; Sreekumar, E.; Leal, G.; et al. Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat. Commun. 2014, 5, 4084. [Google Scholar] [CrossRef]
- Schuffenecker, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.-C.; Lavenir, R.; Pardigon, N.; Reynes, J.-M.; Pettinelli, F.; et al. Genome Microevolution of Chikungunya Viruses Causing the Indian Ocean Outbreak. PLoS Med. 2006, 3, e263. [Google Scholar] [CrossRef]
- Chen, R.; Puri, V.; Fedorova, N.; Lin, D.; Hari, K.L.; Jain, R.; Rodas, J.D.; Das, S.R.; Shabman, R.S.; Weaver, S.C. Comprehensive Genome-Scale Phylogenetic Study Provides New Insights on the Global Expansion of Chikungunya Virus. J. Virol. 2016, 90, 10600–10611. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Quiñonez, J.A.; Ortiz-Alcántara, J.; Fragoso-Fonseca, D.E.; Garcés-Ayala, F.; Escobar-Escamilla, N.; Vázquez-Pichardo, M.; Núñez-León, A.; Torres-Rodríguez, M.D.L.L.; Torres-Longoria, B.; López-Martínez, I.; et al. Complete Genome Sequences of Chikungunya Virus Strains Isolated in Mexico: First Detection of Imported and Autochthonous Cases. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Centro Nacional de Programas Preventivos y Control de Enfermedades. Declaratoria de Emergencia Epidemiológica EE-2-2014. Available online: http://www.cenaprece.salud.gob.mx/programas/interior/emergencias/descargas/pdf/Declaratoria_Emergencia_Chiapas_Chikungunya.pdf (accessed on 20 May 2019).
- Kautz, T.F.; Díaz-González, E.E.; Erasmus, J.H.; Malo-García, I.R.; Langsjoen, R.M.; Patterson, E.I.; Auguste, D.I.; Forrester, N.L.; Sanchez-Casas, R.M.; Hernández-Ávila, M.; et al. Chikungunya Virus as Cause of Febrile Illness Outbreak, Chiapas, Mexico, 2014. Emerg. Infect. Dis. 2015, 21, 2070–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-González, E.E.; Kautz, T.F.; Dorantes-Delgado, A.; Malo-García, I.R.; Laguna-Aguilar, M.; Langsjoen, R.M.; Chen, R.; Auguste, D.I.; Sánchez-Casas, R.M.; Danis-Lozano, R.; et al. First Report of Aedes aegypti Transmission of Chikungunya Virus in the Americas. Am. J. Trop. Med. Hyg. 2015, 93, 1325–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirección General de Epidemiología. Histórico Boletín Epidemiológico. Available online: https://www.gob.mx/salud/acciones-y-programas/historico-boletin-epidemiologico (accessed on 30 April 2019).
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Suchard, M.A.; Lemey, P.; Bernardin, F.J.; Rambaut, A.; Crawford, F.W.; Gray, R.R.; Arinaminpathy, N.; Stramer, S.L.; Busch, M.P.; et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc. Natl. Acad. Sci. USA 2012, 109, 15066–15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, M.W.; Matthews, D.A.; Hiscox, J.A.; Elmore, M.J.; Pollakis, G.; Rambaut, A.; Hewson, R.; García-Dorival, I.; Bore, J.A.; Koundouno, R.; et al. Temporal and spatial analysis of the 2014–2015 Ebola virus outbreak in West Africa. Nature 2015, 524, 97–101. [Google Scholar] [CrossRef]
- Gire, S.K.; Goba, A.; Andersen, K.G.; Sealfon, R.S.G.; Park, D.J.; Kanneh, L.; Jalloh, S.; Momoh, M.; Fullah, M.; Dudas, G.; et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014, 345, 1369–1372. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Kraemer, M.U.G.; Hill, S.C.; Goes de Jesus, J.; Aguiar, R.S.; Iani, F.C.M.; Xavier, J.; Quick, J.; du Plessis, L.; Dellicour, S.; et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018, 361, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Sahu, A.; Das, B.; Das, M.; Patra, A.; Biswal, S.; Kar, S.K.; Hazra, R.K. Genetic characterization of E2 region of Chikungunya virus circulating in Odisha, Eastern India from 2010 to 2011. Infect. Genet. Evol. 2013, 18, 113–124. [Google Scholar] [CrossRef]
- Patil, J.; More, A.; Patil, P.; Jadhav, S.; Newase, P.; Agarwal, M.; Amdekar, S.; Raut, C.G.; Parashar, D.; Cherian, S.S. Genetic characterization of chikungunya viruses isolated during the 2015-2017 outbreaks in different states of India, based on their E1 and E2 genes. Arch. Virol. 2018, 163, 3135–3140. [Google Scholar] [CrossRef]
- Tan, Y.; Pickett, B.E.; Shrivastava, S.; Gresh, L.; Balmaseda, A.; Amedeo, P.; Hu, L.; Puri, V.; Fedorova, N.B.; Halpin, R.A.; et al. Differing epidemiological dynamics of Chikungunya virus in the Americas during the 2014-2015 epidemic. PLoS Negl. Trop. Dis. 2018, 12, e0006670. [Google Scholar] [CrossRef]
- Sahadeo, N.S.D.; Allicock, O.M.; De Salazar, P.M.; Auguste, A.J.; Widen, S.; Olowokure, B.; Gutierrez, C.; Valadere, A.M.; Polson-Edwards, K.; Weaver, S.C.; et al. Understanding the evolution and spread of chikungunya virus in the Americas using complete genome sequences. Virus Evol. 2017, 3, vex010. [Google Scholar] [CrossRef] [Green Version]
- Galán-Huerta, K.; Martínez-Landeros, E.; Delgado-Gallegos, J.; Caballero-Sosa, S.; Malo-García, I.; Fernández-Salas, I.; Ramos-Jiménez, J.; Rivas-Estilla, A. Molecular and Clinical Characterization of Chikungunya Virus Infections in Southeast Mexico. Viruses 2018, 10, 248. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Panella, A.J.; Velez, J.O.; Lambert, A.J.; Campbell, G.L. Chikungunya virus in US travelers returning from India, 2006. Emerg. Infect. Dis. 2007, 13, 764–767. [Google Scholar] [CrossRef]
- Cigarroa-Toledo, N.; Blitvich, B.J.; Cetina-Trejo, R.C.; Talavera-Aguilar, L.G.; Baak-Baak, C.M.; Torres-Chablé, O.M.; Hamid, M.-N.; Friedberg, I.; González-Martinez, P.; Alonzo-Salomon, G.; et al. Chikungunya Virus in Febrile Humans and Aedes aegypti Mosquitoes, Yucatan, Mexico. Emerg. Infect. Dis. 2016, 22, 1804–1807. [Google Scholar] [CrossRef]
- Laredo-Tiscareño, S.V.; Machain-Williams, C.; Rodríguez-Pérez, M.A.; Garza-Hernandez, J.A.; Doria-Cobos, G.L.; Cetina-Trejo, R.C.; Bacab-Cab, L.A.; Tangudu, C.S.; Charles, J.; De Luna-Santillana, E.J.; et al. Arbovirus Surveillance near the Mexico–U.S. Border: Isolation and Sequence Analysis of Chikungunya Virus from Patients with Dengue-like Symptoms in Reynosa, Tamaulipas. Am. J. Trop. Med. Hyg. 2018, 99, 191–194. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. Automated Phylogenetic Detection of Recombination Using a Genetic Algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Griffiths, R.C.; Tavaré, S. Sampling theory for neutral alleles in a varying environment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1994, 344, 403–410. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Tracer v1.6. Available online: http://beast.community/tracer (accessed on 25 July 2019).
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- México en Cifras. Available online: https://www.inegi.org.mx/app/areasgeograficas/?ag=07 (accessed on 20 September2018).
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Stapleford, K.A.; Moratorio, G.; Henningsson, R.; Chen, R.; Matheus, S.; Enfissi, A.; Weissglas-Volkov, D.; Isakov, O.; Blanc, H.; Mounce, B.C.; et al. Whole-Genome Sequencing Analysis from the Chikungunya Virus Caribbean Outbreak Reveals Novel Evolutionary Genomic Elements. PLoS Negl. Trop. Dis. 2016, 10, e0004402. [Google Scholar] [CrossRef]
- Sahadeo, N.; Mohammed, H.; Allicock, O.M.; Auguste, A.J.; Widen, S.G.; Badal, K.; Pulchan, K.; Foster, J.E.; Weaver, S.C.; Carrington, C.V. Molecular Characterisation of Chikungunya Virus Infections in Trinidad and Comparison of Clinical and Laboratory Features with Dengue and Other Acute Febrile Cases. PLoS Negl. Trop. Dis. 2015, 9, e0004199. [Google Scholar] [CrossRef]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef]
- Nunes, M.R.T.; Faria, N.R.; De Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.; De Oliveira, L.F.; Da Silva Azevedo, R.D.S.; Da Silva, D.E.A.; Da Silva, E.V.P.; Da Silva, S.P.; et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015, 13, 102. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Valadere, A.M. Transcontinental Movement of Asian Genotype Chikungunya Virus. Emerg. Infect. Dis. 2014, 20, 1400–1402. [Google Scholar] [CrossRef]
- Lizarazo, E.; Vincenti-Gonzalez, M.; Grillet, M.E.; Bethencourt, S.; Diaz, O.; Ojeda, N.; Ochoa, H.; Rangel, M.A.; Tami, A. Spatial Dynamics of Chikungunya Virus, Venezuela, 2014. Emerg. Infect. Dis. 2019, 25, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Heil, M.L.; Albee, A.; Strauss, J.H.; Kuhn, R.J. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J. Virol. 2001, 75, 6303–6309. [Google Scholar] [CrossRef]
- Meyer, W.J.; Gidwitz, S.; Ayers, V.K.; Schoepp, R.J.; Johnston, R.E. Conformational alteration of Sindbis virion glycoproteins induced by heat, reducing agents, or low pH. J. Virol. 1992, 66, 3504–3513. [Google Scholar] [PubMed]
- Meyer, W.J.; Johnston, R.E. Structural rearrangement of infecting Sindbis virions at the cell surface: Mapping of newly accessible epitopes. J. Virol. 1993, 67, 5117–5125. [Google Scholar]
- Byrd, E.A.; Kielian, M. An Alphavirus E2 Membrane-Proximal Domain Promotes Envelope Protein Lateral Interactions and Virus Budding. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Data Set | Codon Position | Significance | Mutation | Sequences |
|---|---|---|---|---|---|
| SLAC | Asian | - | - | - | - |
| Chiapas | - | - | - | - | |
| FEL | Asian | 221 | 0.031 b | K → G | Thailand 1958 (HM045810) |
| K → R | Thailand 1996 (KX262987) | ||||
| Philippines 2011 (KU561459) | |||||
| Curacao 2015 (KU355832) | |||||
| Nicaragua 2015 (KY703948) | |||||
| Nicaragua 2015 (KY703966) | |||||
| Puerto Rico 2015 (MF001515) | |||||
| TA689 2015 (MH481882) | |||||
| Cuba 2016 (LC259092) | |||||
| Chiapas | - | - | - | - | |
| FUBAR | Asian | 221 | 0.991 c | K → G | Thailand 1958 (HM045810) |
| K → R | Thailand 1996 (KX262987) | ||||
| Philippines 2011 (KU561459) | |||||
| Curacao 2015 (KU355832) | |||||
| Nicaragua 2015 (KY703948) | |||||
| Nicaragua 2015 (KY703966) | |||||
| Puerto Rico 2015 (MF001515) | |||||
| TA689 2015 (MH481882) | |||||
| Cuba 2016 (LC259092) | |||||
| Chiapas | - | - | - | - | |
| MEME | Asian | 221 | 0.01 b | K → G | Thailand 1958 (HM045810) |
| K → R | Thailand 1996 (KX262987) | ||||
| Philippines 2011 (KU561459) | |||||
| Curacao 2015 (KU355832) | |||||
| Nicaragua 2015 (KY703948) | |||||
| Nicaragua 2015 (KY703966) | |||||
| Puerto Rico 2015 (MF001515) | |||||
| TA689 2015 (MH481882) | |||||
| Cuba 2016 (LC259092) | |||||
| 353 | <0.001 b | H → A | Yucatán 2015 (KU295119 – 26, 28–30) | ||
| H → E | Yucatán 2015 (KU295127) | ||||
| Chiapas | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galan-Huerta, K.A.; Zomosa-Signoret, V.C.; Vidaltamayo, R.; Caballero-Sosa, S.; Fernández-Salas, I.; Ramos-Jiménez, J.; Rivas-Estilla, A.M. Genetic Variability of Chikungunya Virus in Southern Mexico. Viruses 2019, 11, 714. https://doi.org/10.3390/v11080714
Galan-Huerta KA, Zomosa-Signoret VC, Vidaltamayo R, Caballero-Sosa S, Fernández-Salas I, Ramos-Jiménez J, Rivas-Estilla AM. Genetic Variability of Chikungunya Virus in Southern Mexico. Viruses. 2019; 11(8):714. https://doi.org/10.3390/v11080714
Chicago/Turabian StyleGalan-Huerta, Kame A., Viviana C. Zomosa-Signoret, Román Vidaltamayo, Sandra Caballero-Sosa, Ildefonso Fernández-Salas, Javier Ramos-Jiménez, and Ana M. Rivas-Estilla. 2019. "Genetic Variability of Chikungunya Virus in Southern Mexico" Viruses 11, no. 8: 714. https://doi.org/10.3390/v11080714
APA StyleGalan-Huerta, K. A., Zomosa-Signoret, V. C., Vidaltamayo, R., Caballero-Sosa, S., Fernández-Salas, I., Ramos-Jiménez, J., & Rivas-Estilla, A. M. (2019). Genetic Variability of Chikungunya Virus in Southern Mexico. Viruses, 11(8), 714. https://doi.org/10.3390/v11080714