Combinatorial Avidity Selection of Mosaic Landscape Phages Targeted at Breast Cancer Cells—An Alternative Mechanism of Directed Molecular Evolution

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Landscape Phage Display Library
2.3. Selection of Breast Cancer Cell-Specific Phages
2.3.1. Depletion and First Round Selection
2.3.2. Washing and Sublibrary Generation
2.3.3. Second, Third and Fourth Rounds
2.3.4. Sequencing of Sublibraries
2.4. Computational Analysis
2.5. Specificity and Selectivity of Phages
2.6. Immunofluorescence Analysis of Phages
3. Results
3.1. Selection of Breast Cancer Cell-Specific Landscape Phages
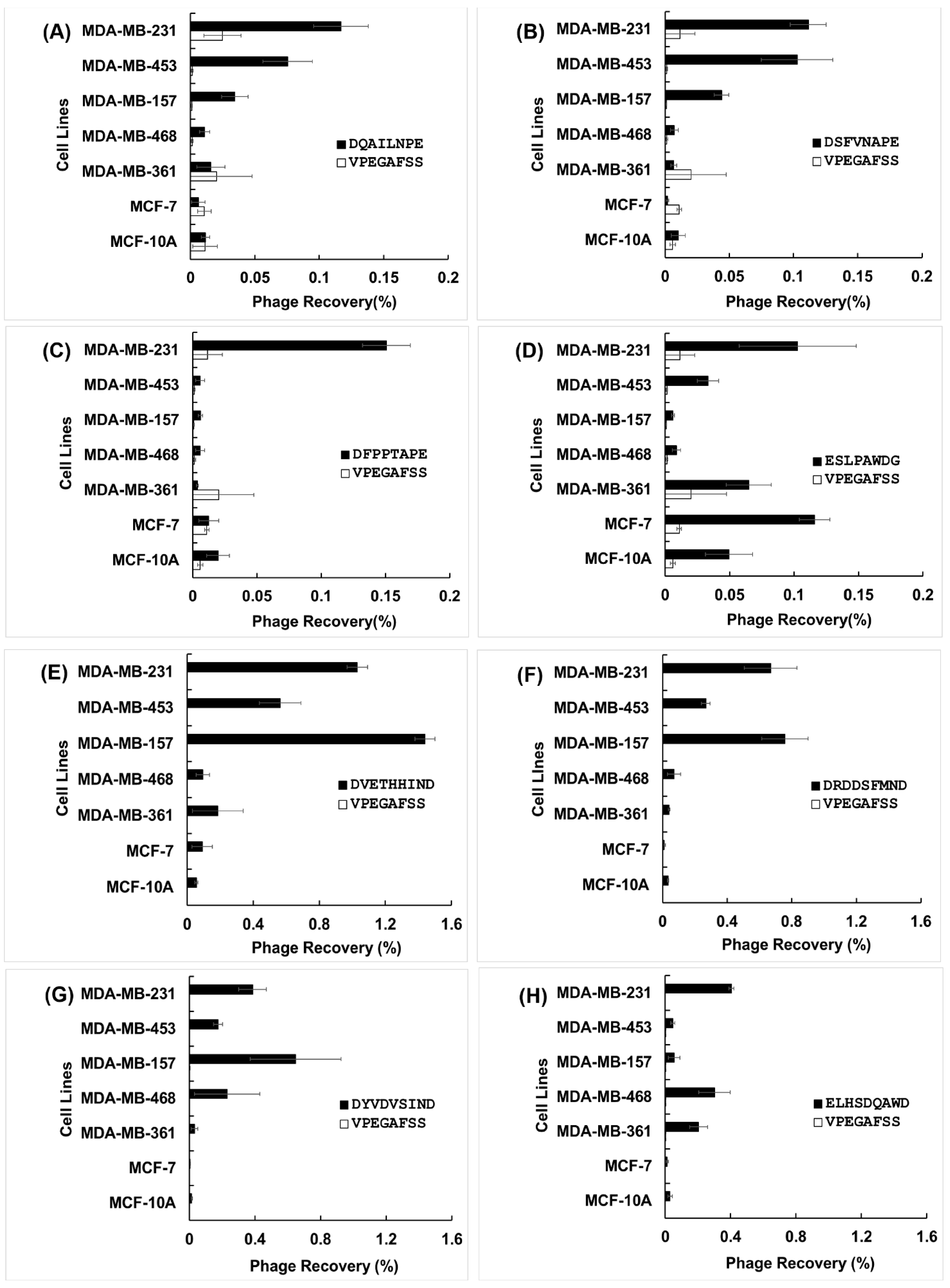
3.2. Specificity and Selectivity of Phages Towards Breast Cancer Cells
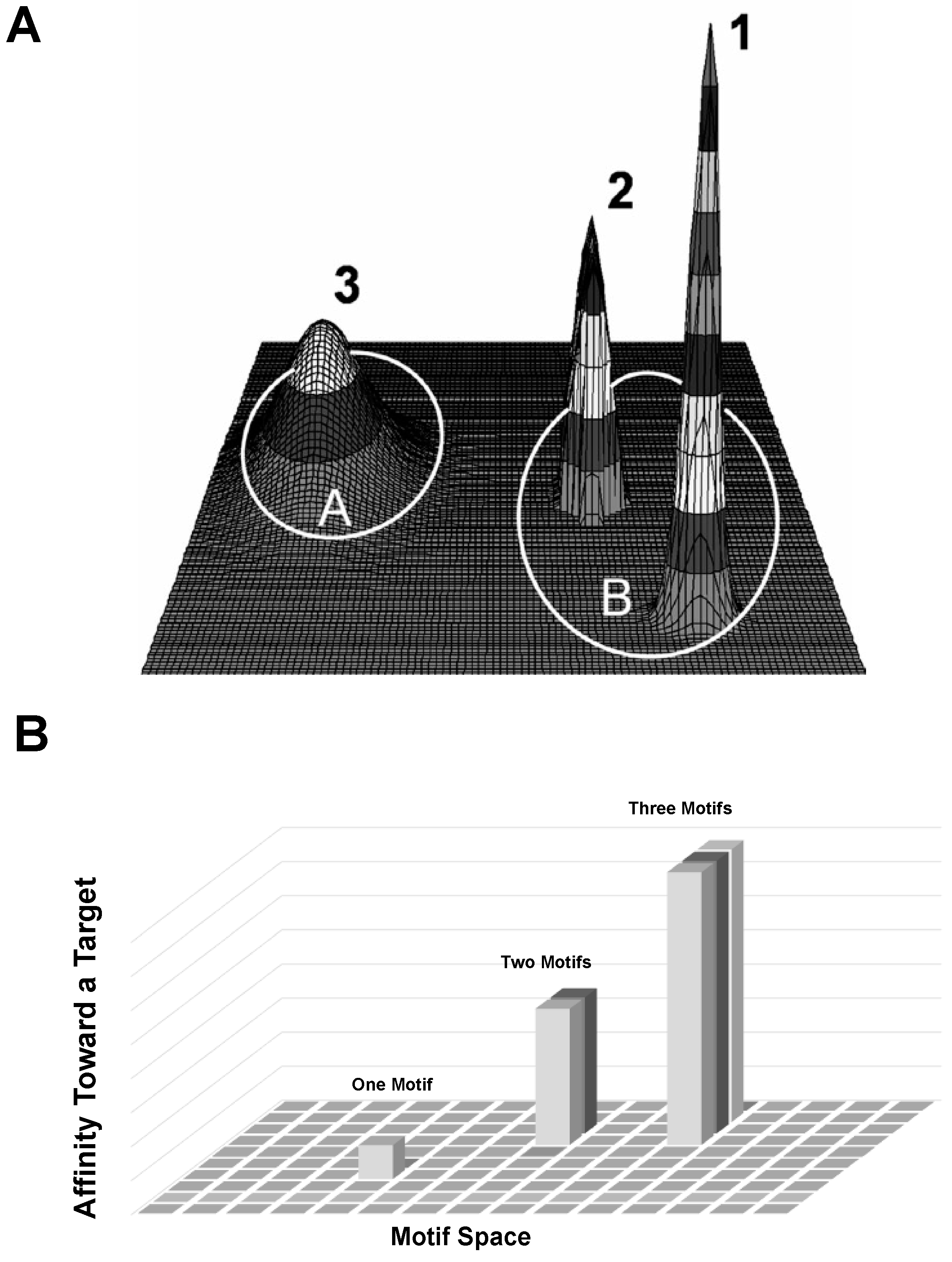
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Petrenko, V.A. Autonomous self-navigating drug-delivery vehicles: From science fiction to reality. Ther. Deliv. 2017, 8, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.W.; de Avila, B.E.F.; Zhang, L.F.; Wang, J. Targeting and isolation of cancer cells using micro/nanomotors. Adv. Drug Deliv. Rev. 2018, 125, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Clergeaud, G.; Andresen, T.L.; Boisen, A. Micromotors for drug delivery in vivo: The road ahead. Adv. Drug Deliv. Rev. 2019, 138, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.B.; Gu, Z.J.; An, H.W.; Chen, C.Y.; Chen, J.; Cui, R.; Chen, S.Q.; Chen, W.H.; Chen, X.S.; Chen, X.Y.; et al. Precise nanomedicine for intelligent therapy of cancer. Sci. China Chem. 2018, 61, 1503–1552. [Google Scholar] [CrossRef]
- Schuerle, S.; Soleimany, A.P.; Yeh, T.; Anand, G.M.; Haberli, M.; Fleming, H.E.; Mirkhani, N.; Qiu, F.; Hauert, S.; Wang, X.; et al. Synthetic and living micropropellers for convection-enhanced nanoparticle transport. Sci. Adv. 2019, 5, 10. [Google Scholar] [CrossRef]
- Abendroth, J.M.; Bushuyev, O.S.; Weiss, P.S.; Barrett, C.J. Controlling motion at the nanoscale: Rise of the molecular machines. ACS Nano 2015, 9, 7746–7768. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, V.A. Landscape phage: Evolution from phage display to nanobiotechnology. Viruses 2018, 10, 311. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Neduva, V.; Russell, R.B. Linear motifs: Evolutionary interaction switches. FEBS Lett. 2005, 579, 3342–3345. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Giver, L.; Shao, Z.; Affholter, J.A.; Arnold, F.H. Molecular evolution by staggered extension process (step) in vitro recombination. Nat. Biotechnol. 1998, 16, 258–261. [Google Scholar] [CrossRef]
- Davey, N.E.; Cyert, M.S.; Moses, A.M. Short linear motifs—Ex nihilo evolution of protein regulation. Cell Commun. Signal. 2015, 13. [Google Scholar] [CrossRef]
- Gouw, M.; Michael, S.; Samano-Sanchez, H.; Kumar, M.; Zeke, A.; Lang, B.; Bely, B.; Chemes, L.B.; Davey, N.E.; Deng, Z.; et al. The eukaryotic linear motif resource—2018 update. Nucleic Acids Res. 2018, 46, D428–D434. [Google Scholar] [CrossRef]
- Li, X.H.; Babu, M.M. Human diseases from gain-of-function mutations in disordered protein regions. Cell 2018, 175, 40–42. [Google Scholar] [CrossRef]
- Gross, A.L.; Gillespie, J.W.; Petrenko, V.A. Promiscuous tumor targeting phage proteins. Protein Eng. Des. Sel. 2016, 29, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. Biosyst. 2012, 8, 268–281. [Google Scholar] [CrossRef]
- Diella, F.; Haslam, N.; Chica, C.; Budd, A.; Michael, S.; Brown, N.P.; Trave, G.; Gibson, T.J. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front. Biosci. Landmrk. 2008, 13, 6580–6603. [Google Scholar] [CrossRef]
- Van Roey, K.; Gibson, T.J.; Davey, N.E. Motif switches: Decision-making in cell regulation. Curr. Opin. Struct. Biol. 2012, 22, 378–385. [Google Scholar] [CrossRef]
- Van Roey, K.; Dinkel, H.; Weatheritt, R.J.; Gibson, T.J.; Davey, N.E. The switches. Elm resource: A compendium of conditional regulatory interaction interfaces. Sci. Signal. 2013, 6, rs7. [Google Scholar] [CrossRef]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Davey, N.E.; Seo, M.H.; Yadav, V.K.; Jeon, J.; Nim, S.; Krystkowiak, I.; Blikstad, C.; Dong, D.; Markova, N.; Kim, P.M.; et al. Discovery of short linear motif-mediated interactions through phage display of intrinsically disordered regions of the human proteome. FEBS J. 2017, 284, 485–498. [Google Scholar] [CrossRef]
- Knez, K.; Noppe, W.; Geukens, N.; Janssen, K.P.; Spasic, D.; Heyligen, J.; Vriens, K.; Thevissen, K.; Cammue, B.P.; Petrenko, V.; et al. Affinity comparison of p3 and p8 peptide displaying bacteriophages using surface plasmon resonance. Anal. Chem. 2013, 85, 10075–10082. [Google Scholar] [CrossRef]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, V.A.; Smith, G.P.; Gong, X.; Quinn, T. A library of organic landscapes on filamentous phage. Protein Eng. 1996, 9, 797–801. [Google Scholar] [CrossRef]
- Kuzmicheva, G.A.; Jayanna, P.K.; Sorokulova, I.B.; Petrenko, V.A. Diversity and censoring of landscape phage libraries. Protein Eng. Des. Sel. 2009, 22, 9–18. [Google Scholar] [CrossRef]
- Brigati, J.R.; Samoylova, T.I.; Jayanna, P.K.; Petrenko, V.A. Phage display for generating peptide reagents. Curr. Protoc. Protein Sci. 2008, 51, 18–19. [Google Scholar]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. Meme suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- He, B.; Chai, G.; Duan, Y.; Yan, Z.; Qiu, L.; Zhang, H.; Liu, Z.; He, Q.; Han, K.; Ru, B.; et al. Bdb: Biopanning data bank. Nucleic Acids Res. 2016, 44, D1127–D1132. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. Interpro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef]
- Richardson, L.J.; Rawlings, N.D.; Salazar, G.A.; Almeida, A.; Haft, D.R.; Ducq, G.; Sutton, G.G.; Finn, R.D. Genome properties in 2019: A new companion database to interpro for the inference of complete functional attributes. Nucleic Acids Res. 2019, 47, D564–D572. [Google Scholar] [CrossRef]
- Jayanna, P.K.; Bedi, D.; Deinnocentes, P.; Bird, R.C.; Petrenko, V.A. Landscape phage ligands for pc3 prostate carcinoma cells. Protein Eng. Des. Sel. 2010, 23, 423–430. [Google Scholar] [CrossRef]
- Fagbohun, O.A.; Bedi, D.; Grabchenko, N.I.; Deinnocentes, P.A.; Bird, R.C.; Petrenko, V.A. Landscape phages and their fusion proteins targeted to breast cancer cells. Protein Eng. Des. Sel. 2012, 25, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, V.A.; Smith, G.P. Phages from landscape libraries as substitute antibodies. Protein Eng. 2000, 13, 589–592. [Google Scholar] [CrossRef] [Green Version]
- Fagbohun, O.A.; Kazmierczak, R.A.; Petrenko, V.A.; Eisenstark, A. Metastatic prostate cancer cell-specific phage-like particles as a targeted gene-delivery system. J. Nanobiotechnol. 2013, 11, 31. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A.; Matthews, L.J. Cross-linked filamentous phage as an affinity matrix. J. Immunol. Methods 1998, 215, 151–161. [Google Scholar] [CrossRef]
- Cailleau, R.; Young, R.; Olive, M.; Reeves, W.J., Jr. Breast tumor cell lines from pleural effusions. J. Natl. Cancer Inst. 1974, 53, 661–674. [Google Scholar] [CrossRef]
- Gillespie, J.W.; Wei, L.; Petrenko, V.A. Selection of lung cancer-specific landscape phage for targeted drug delivery. Comb. Chem. High Throughput Screen. 2016, 19, 412–422. [Google Scholar] [CrossRef]
- Teyra, J.; Huang, H.; Jain, S.; Guan, X.; Dong, A.; Liu, Y.; Tempel, W.; Min, J.; Tong, Y.; Kim, P.M.; et al. Comprehensive analysis of the human sh3 domain family reveals a wide variety of non-canonical specificities. Structure 2017, 25, 1598–1610. [Google Scholar] [CrossRef]
- Saksela, K.; Permi, P. Sh3 domain ligand binding: What’s the consensus and where’s the specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef]
- Kurochkina, N.; Guha, U. Sh3 domains: Modules of protein-protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef]
- Stoll, R.; Bosserhoff, A. Extracellular sh3 domain containing proteins—Features of a new protein family. Curr. Protein Pept. Sci. 2008, 9, 221–226. [Google Scholar] [CrossRef]
- Pornillos, O.; Alam, S.L.; Davis, D.R.; Sundquist, W.I. Structure of the tsg101 uev domain in complex with the ptap motif of the hiv-1 p6 protein. Nat. Struct. Biol. 2002, 9, 812–817. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Schubert, H.L.; Kelly, B.N.; Hill, G.C.; Holton, J.M.; Hill, C.P. Ubiquitin recognition by the human tsg101 protein. Mol. Cell 2004, 13, 783–789. [Google Scholar] [CrossRef]
- Ahmed, I.; Akram, Z.; Iqbal, H.M.N.; Munn, A.L. The regulation of endosomal sorting complex required for transport and accessory proteins in multivesicular body sorting and enveloped viral budding—An overview. Int. J. Biol. Macromol. 2019, 127, 1–11. [Google Scholar] [CrossRef]
- Dussupt, V.; Javid, M.P.; Abou-Jaoude, G.; Jadwin, J.A.; de La Cruz, J.; Nagashima, K.; Bouamr, F. The nucleocapsid region of hiv-1 gag cooperates with the ptap and lypxnl late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 2009, 5, e1000339. [Google Scholar] [CrossRef]
- Irie, T.; Harty, R.N. L-domain flanking sequences are important for host interactions and efficient budding of vesicular stomatitis virus recombinants. J. Virol. 2005, 79, 12617–12622. [Google Scholar] [CrossRef]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping motifs (ptap and ppey) within the ebola virus vp40 protein function independently as late budding domains: Involvement of host proteins tsg101 and vps-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef]
- Ivanenkov, V.V.; Menon, A.G. Peptide-mediated transcytosis of phage display vectors in mdck cells. Biochem. Biophys. Res. Commun. 2000, 276, 251–257. [Google Scholar] [CrossRef]
- Park, J.E.; Soung, N.K.; Johmura, Y.; Kang, Y.H.; Liao, C.; Lee, K.H.; Park, C.H.; Nicklaus, M.C.; Lee, K.S. Polo-box domain: A versatile mediator of polo-like kinase function. Cell Mol. Life Sci. 2010, 67, 1957–1970. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jang, C.; Lee, K.A. Polo-like kinases (plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev. Reprod. 2014, 18, 65–71. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, A.R.; Sharma, G.; Chakraborty, C.; Kim, J. Plk-1: Angel or devil for cell cycle progression. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 190–203. [Google Scholar] [CrossRef]
- Durocher, D.; Jackson, S.P. The fha domain. FEBS Lett. 2002, 513, 58–66. [Google Scholar] [CrossRef]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2010, 15, 39–48. [Google Scholar] [CrossRef]
- Ozbabacan, S.E.A.; Engin, H.B.; Gursoy, A.; Keskin, O. Transient protein-protein interactions. Protein Eng. Des. Sel. 2011, 24, 635–648. [Google Scholar] [CrossRef]
- Yu, J.N.; Smith, G.P. Affinity maturation of phage-displayed peptide ligands. Method Enzymol. 1996, 267, 3–27. [Google Scholar]
- Petrenko, V.A.; Gillespie, J.W. Paradigm shift in bacteriophage-mediated delivery of anticancer drugs: From targeted magic bullets to self-navigated magic missiles. Expert Opin. Drug Deliv. 2017, 14, 373–384. [Google Scholar] [CrossRef]
- Kim, E.S. Directed evolution: A historical exploration into an evolutionary experimental system of nanobiotechnology, 1965–2006. Minerva 2008, 46, 463–484. [Google Scholar] [CrossRef]
- Marvin, D.A.; Hale, R.D.; Nave, C.; Helmer-Citterich, M. Molecular models and structural comparisons of native and mutant class i filamentous bacteriophages ff (fd, f1, m13), if1 and ike. J. Mol. Biol. 1994, 235, 260–286. [Google Scholar] [CrossRef]
- Marvin, D.A.; Symmons, M.F.; Straus, S.K. Structure and assembly of filamentous bacteriophages. Prog. Biophys. Mol. Biol. 2014, 114, 80–122. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strong | Medium | Weak |
|---|---|---|
| DTFAKSMQ | DTFAKSAS | ETFAASRR |
| ETFAKMSQ | DTFAKSNA | ETFASRSS |
| ETFAKMTQ | ETFARSQS | ETFAASNR |
| DTFAKMAQ | DTFARQNA | ETFASTRS |
| ETFAKSNA | ||
| DTFARTQS | ||
| ETFARSNS | ||
| DTFARSSS | ||
| ETFASRSQ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrenko, V.A.; Gillespie, J.W.; Xu, H.; O’Dell, T.; De Plano, L.M. Combinatorial Avidity Selection of Mosaic Landscape Phages Targeted at Breast Cancer Cells—An Alternative Mechanism of Directed Molecular Evolution. Viruses 2019, 11, 785. https://doi.org/10.3390/v11090785
Petrenko VA, Gillespie JW, Xu H, O’Dell T, De Plano LM. Combinatorial Avidity Selection of Mosaic Landscape Phages Targeted at Breast Cancer Cells—An Alternative Mechanism of Directed Molecular Evolution. Viruses. 2019; 11(9):785. https://doi.org/10.3390/v11090785
Chicago/Turabian StylePetrenko, Valery A., James W. Gillespie, Hai Xu, Tiffany O’Dell, and Laura M. De Plano. 2019. "Combinatorial Avidity Selection of Mosaic Landscape Phages Targeted at Breast Cancer Cells—An Alternative Mechanism of Directed Molecular Evolution" Viruses 11, no. 9: 785. https://doi.org/10.3390/v11090785
APA StylePetrenko, V. A., Gillespie, J. W., Xu, H., O’Dell, T., & De Plano, L. M. (2019). Combinatorial Avidity Selection of Mosaic Landscape Phages Targeted at Breast Cancer Cells—An Alternative Mechanism of Directed Molecular Evolution. Viruses, 11(9), 785. https://doi.org/10.3390/v11090785